


As doenças de armazenamento lisossomal são um grupo de perturbações metabólicas causadas por mutações genéticas nas enzimas responsáveis pela função lisossomal normal. A disfunção dos processos enzimáticos causa uma acumulação de metabolitos não digeridos, o que resulta em morte celular. Os principais grupos de doenças de armazenamento lisossomal incluem esfingolipidoses, oligossacaridoses e mucolipidoses Mucolipidoses A group of inherited metabolic diseases characterized by the accumulation of excessive amounts of acid mucopolysaccharides, sphingolipids, and/or glycolipids in visceral and mesenchymal cells. Abnormal amounts of sphingolipids or glycolipids are present in neural tissue. Intellectual disability and skeletal changes, most notably dysostosis multiplex, occur frequently. Overview of Lysosomal Storage Diseases. Os subgrupos das doenças principais têm mecanismos subjacentes e manifestações clínicas diferentes.
Last updated: Apr 24, 2025
As doenças de armazenamento lisossomal são patologias metabólicas raras causadas por mutações genéticas de enzimas lisossomais, que levam ao metabolismo disfuncional e à acumulação de glicosaminoglicanos, glicoproteínas ou glicolípidos.
As perturbações são causadas por uma deficiência de uma hidrolase lisossomal específica ou de enzimas necessárias para a função lisossomal:
As perturbações são consideradas como grupos de perturbações hereditárias individualmente raras do metabolismo intracelular. Das 40 perturbações classificadas, 15 são responsáveis pela maioria dos casos. A categorização é por metabolitos intermediários acumulados:
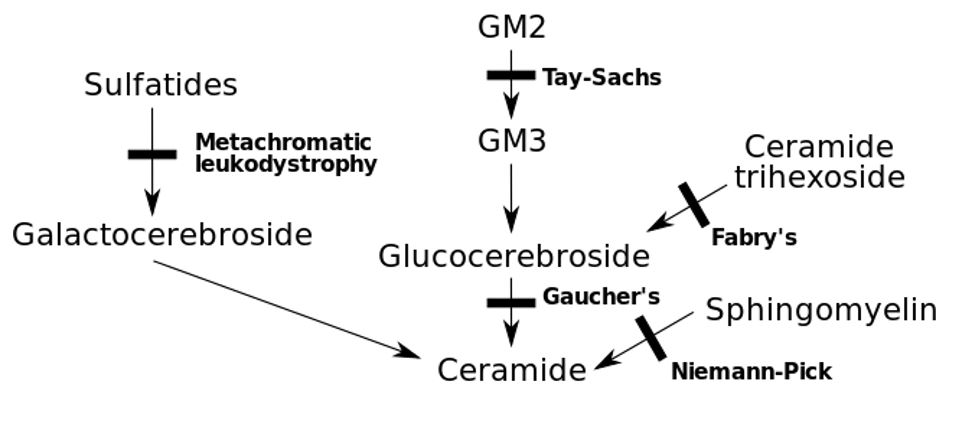
Erros inatos do metabolismo com o défice genético associado
Imagem: “Inborn errors of metabolism” by Huckfinne. Licença: Public Domain| Grupo | Subgrupo | Descrição |
|---|---|---|
| Esfingolipidoses | GM2-gangliosidose |
|
| GM1-gangliosidose | Causada por uma mutação no gene Gene A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. Basic Terms of Genetics GLB1 que codifica para a β-galactosidase-1 | |
| Doença de Gaucher |
|
|
| Doença de Fabry |
|
|
| Leucodistrofia metacromática | Deficiência na atividade da arilsulfatase A | |
| Doença de Krabbe |
|
|
| Lipogranulomatose disseminada (doença de Farber) |
|
|
| Doença de Niemann-Pick |
|
|
| Oligossacaridoses | Sialidose |
|
| Galactosialidose |
|
|
| Fucosidose |
|
|
| Manosidose | 2 tipos:
|
|
| Mucolipidoses Mucolipidoses A group of inherited metabolic diseases characterized by the accumulation of excessive amounts of acid mucopolysaccharides, sphingolipids, and/or glycolipids in visceral and mesenchymal cells. Abnormal amounts of sphingolipids or glycolipids are present in neural tissue. Intellectual disability and skeletal changes, most notably dysostosis multiplex, occur frequently. Overview of Lysosomal Storage Diseases | Doença de I-cell |
|
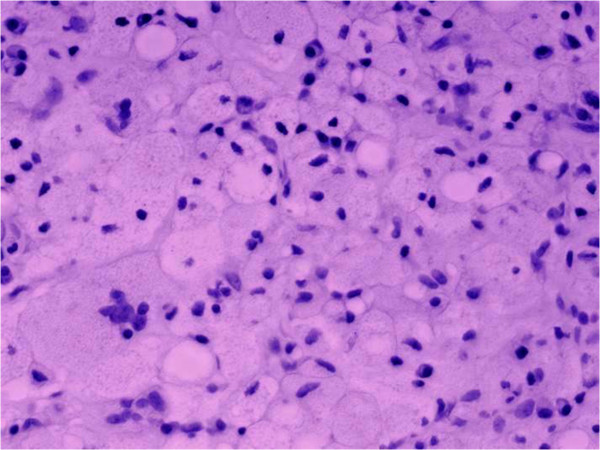
A doença de Gaucher leva à necrose óssea:
O tecido conjuntivo está infiltrado com várias células de Gaucher (células reticuloendoteliais carregadas de lípidos, vacuoladas, com citoplasma granular expandido e núcleos arredondados e deslocados).
A apresentação é variável dependendo da etiologia da perturbação de armazenamento lisossomal e pode ocorrer logo após o nascimento ou mais MAIS Androgen Insensitivity Syndrome tarde na idade adulta:
| Grupo | Subgrupo | Sinais e sintomas |
|---|---|---|
| Esfingolipidoses | GM2-gangliosidose |
|
| GM1-gangliosidose |
|
|
| Doença de Gaucher |
|
|
| Doença de Fabry |
|
|
| Leucodistrofia metacromática |
|
|
| Doença de Krabbe |
|
|
| Doença de Niemann-Pick |
|
|
| Oligossacaridoses | Sialidose |
|
| Galactosialidose |
|
|
| Fucosidose |
|
|
| Mucolipidoses Mucolipidoses A group of inherited metabolic diseases characterized by the accumulation of excessive amounts of acid mucopolysaccharides, sphingolipids, and/or glycolipids in visceral and mesenchymal cells. Abnormal amounts of sphingolipids or glycolipids are present in neural tissue. Intellectual disability and skeletal changes, most notably dysostosis multiplex, occur frequently. Overview of Lysosomal Storage Diseases | Doença de I-cell |
|
Os exames são baseados na doença de armazenamento lisossomal específica, mas alguns princípios gerais são importantes para entender:
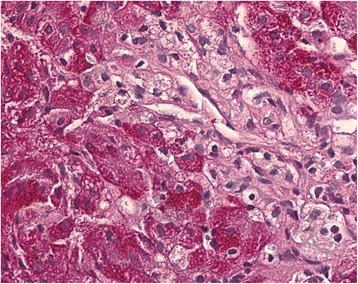
Uma célula de Niemann-Pick de uma amostra de fígado que motras células de Kupffer inchadas com citoplasma espumoso, típico para doença de Niemann-Pick tipo C
Imagem: “Histopathological liver biopsy findings” by Degtyareva AV, Mikhailova SV, Zakharova EY, Tumanova EL, Puchkova AA. Licença: CC BY 4.0O tratamento depende da perturbação específica. Os princípios gerais do tratamento incluem:
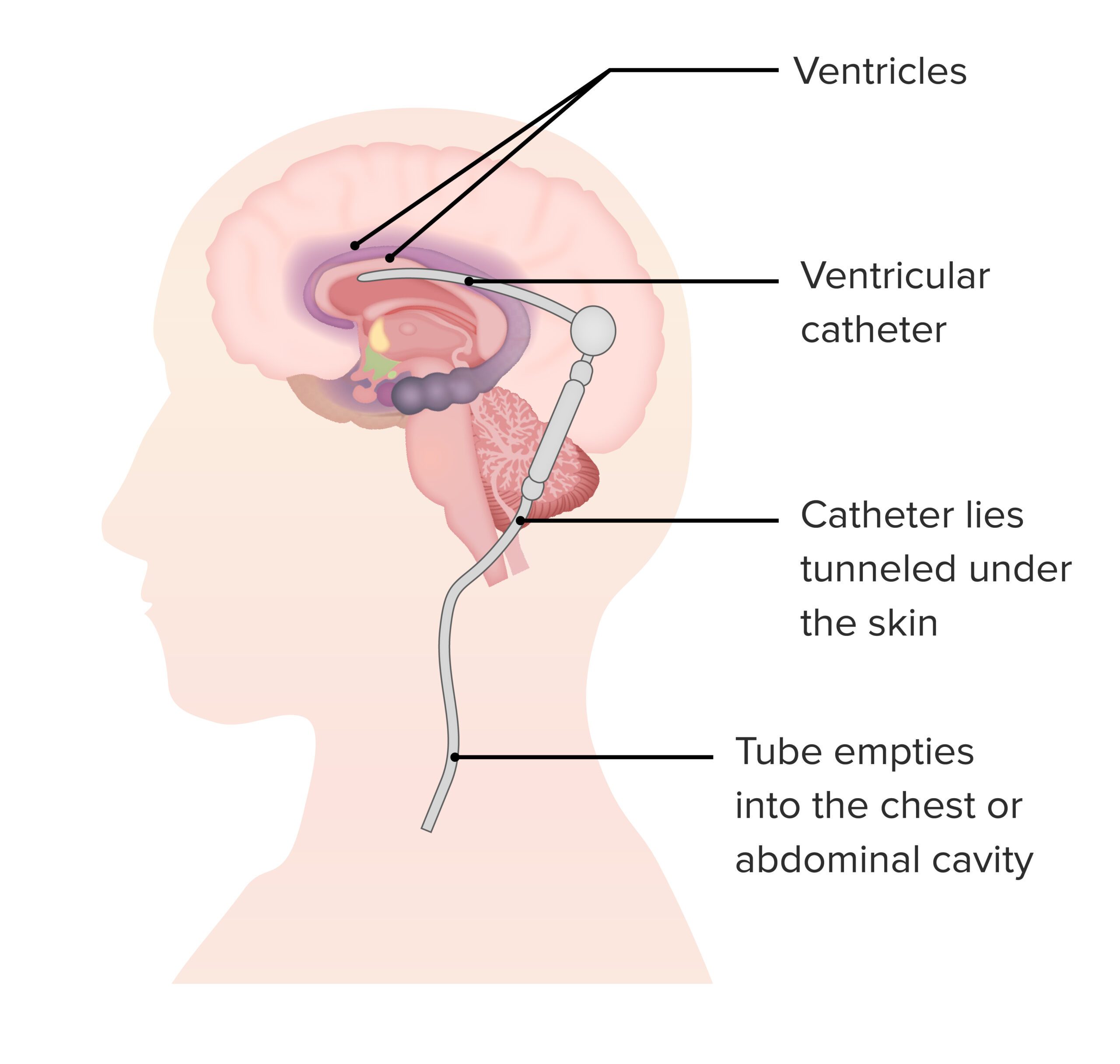
Pode ser necessário um shunt para indivíduos com hidrocefalia por uma perturbação de armazenamento lisossomal.
Imagem por Lecturio.Embora as perturbações de armazenamento lisossomal incluam um amplo espetro de doenças, várias outras patologias podem-se sobrepor às perturbações. Algumas patologias são subgrupos no grupo geral de perturbações de armazenamento lisossomal:
