


As proteínas correspondem a um dos três principais macronutrientes do corpo e são sintetizadas a partir de elementos de construção individuais designados aminoácidos (AAs). Os aminoácidos unem-se através de ligações peptídicas, que ligam a extremidade amino de um AA AA Amyloidosis à extremidade carboxi do AA AA Amyloidosis seguinte, dando origem à estrutura primária da proteína. Posteriormente, a cadeia de AAs sofre novas alterações, dando origem a estruturas tridimensionais complexas. As proteínas apresentam uma grande diversidade de funções, incluindo funções catalíticas, estruturais, de regulação, de transporte, armazenamento e imunológicas. Estas são digeridas por ação das proteases Proteases Proteins and Peptides e das peptidases secretadas pelo estômago e pelo pâncreas e são posteriormente absorvidos como AAs individuais no intestino delgado através de transportadores especializados. Existem inúmeras patologias relacionadas com anomalias das proteínas, entre as quais perturbações relacionadas com as enzimas, recetores, canais presentes na membrana, hormonas, acumulação de proteínas e patologias autoimunes.
Last updated: Dec 15, 2025
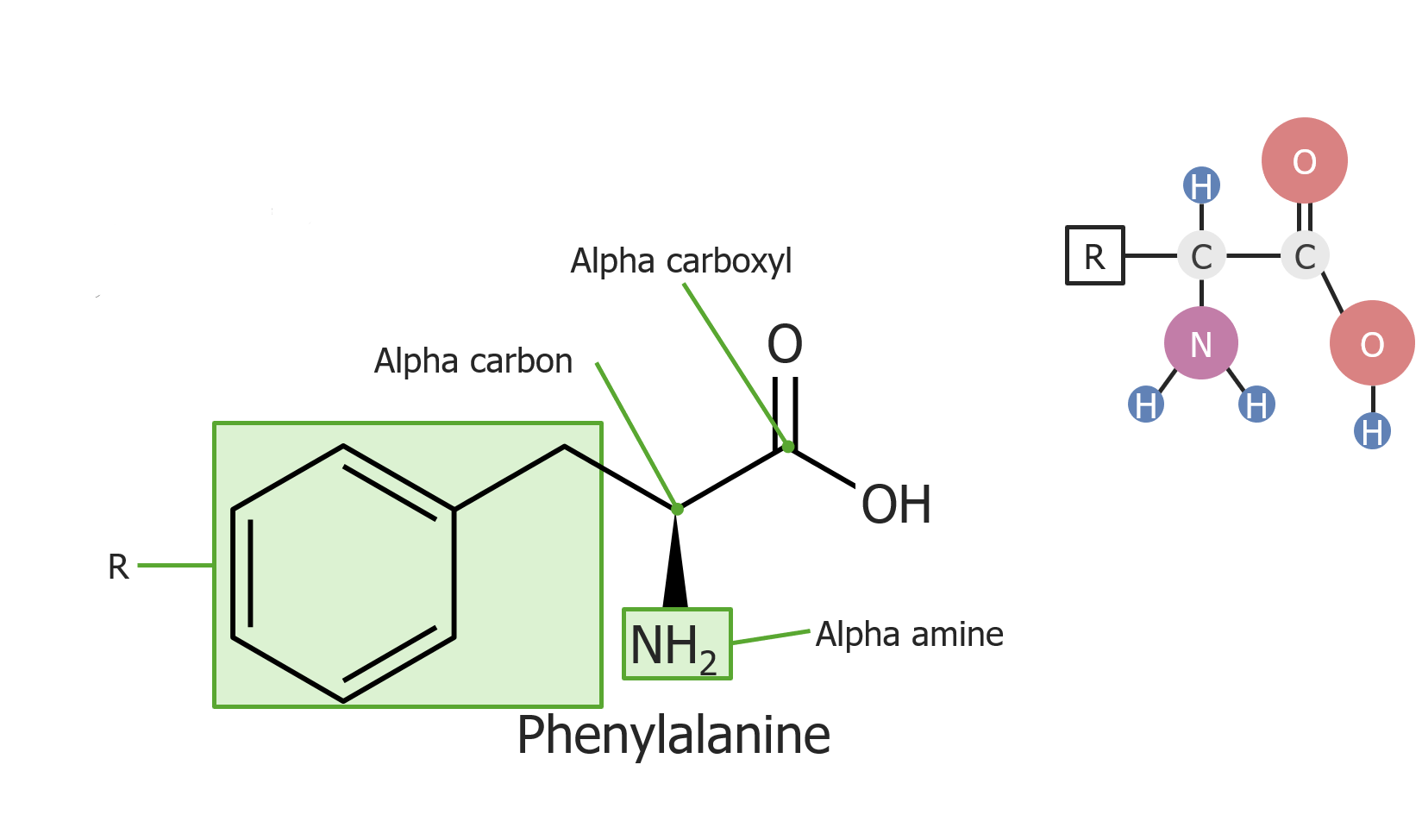
Exemplo do aminoácido fenilalanina
Imagem por Lecturio.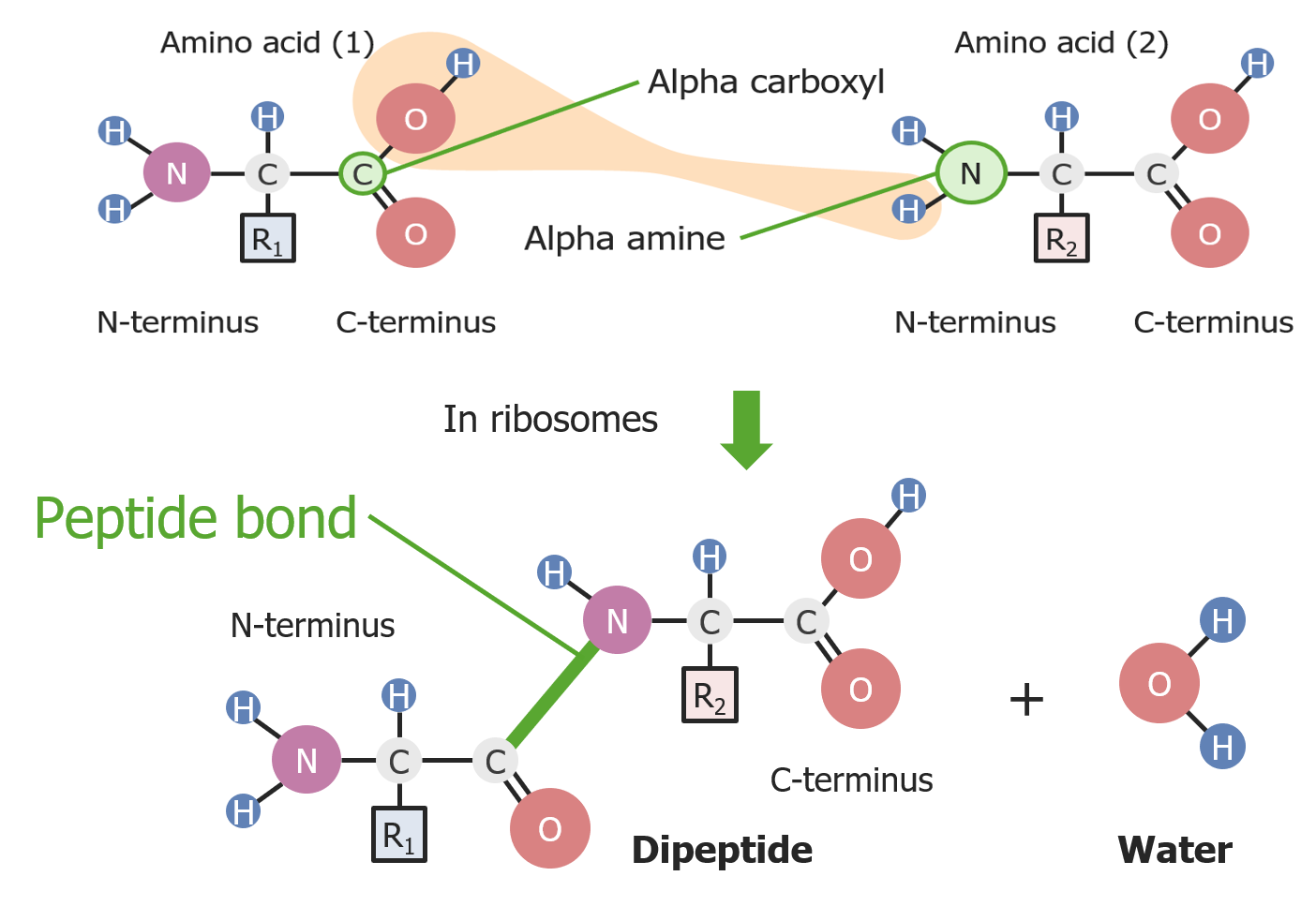
Formação de uma ligação peptídica entre 2 aminoácidos
Imagem por Lecturio.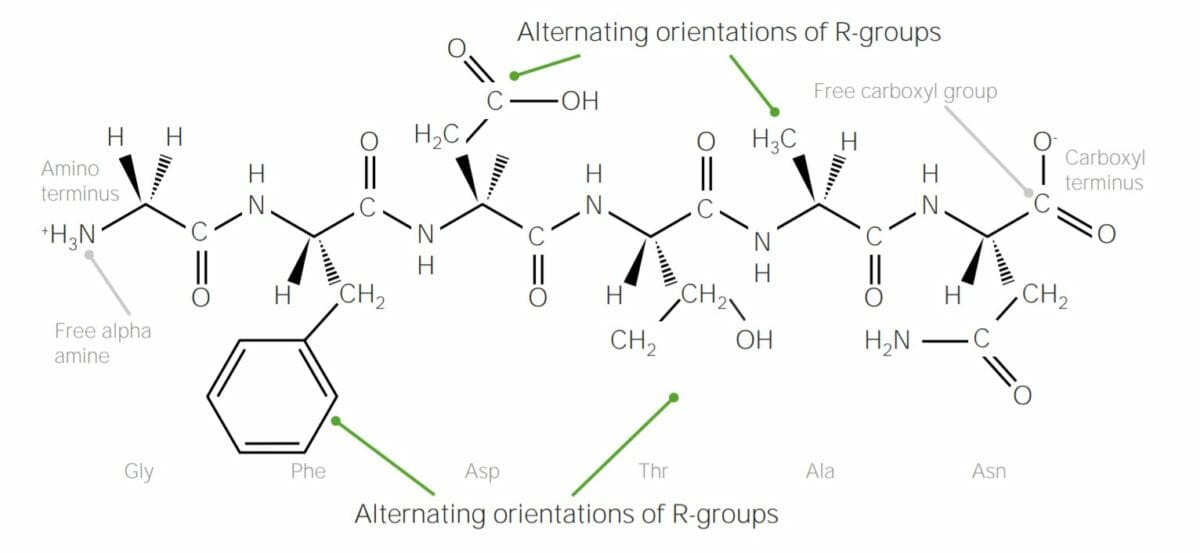
Exemplo de um polipeptídeo que contem 6 aminoácidos, unidos por ligações peptídicas:
Nesta imagem é possível observar que os grupos R de cada aminoácido alternam de “lados” na cadeia polipeptídica; isto ocorre devido à forma trans das ligações peptídicas.
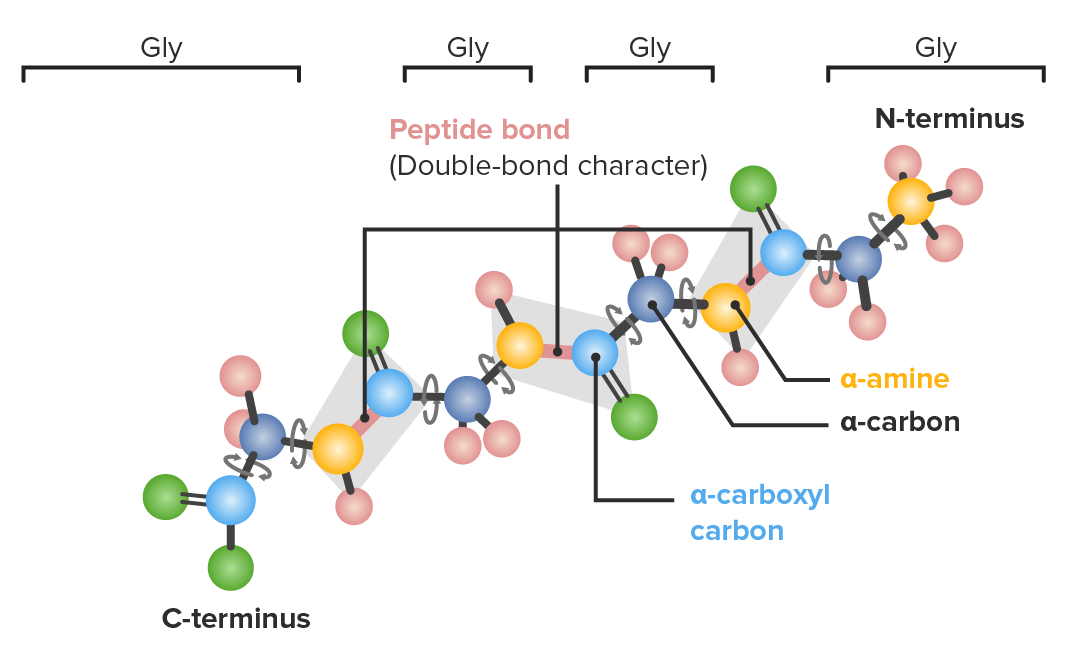
A imagem apresentada representa um exemplo de um polipeptídeo constituído por 4 aminoácidos de glicina (gli), no qual é possível observar as ligações que apresentam capacidade rotacional:
Azul escuro: α-carbonos
Azul claro: carbonos carboxílicos
Amarelo: nitrogénio
Verde: oxigénio
Rosa: hidrogénio
Existem 4 níveis de estrutura proteica; muitas vezes, designados, como o “folding” proteico. Os níveis de estrutura são, a primária, secundária, terciária e quaternária. Para ocorrer um “folding” adequado é necessária a ajuda das proteínas “chaperones“
Estrutura primária:
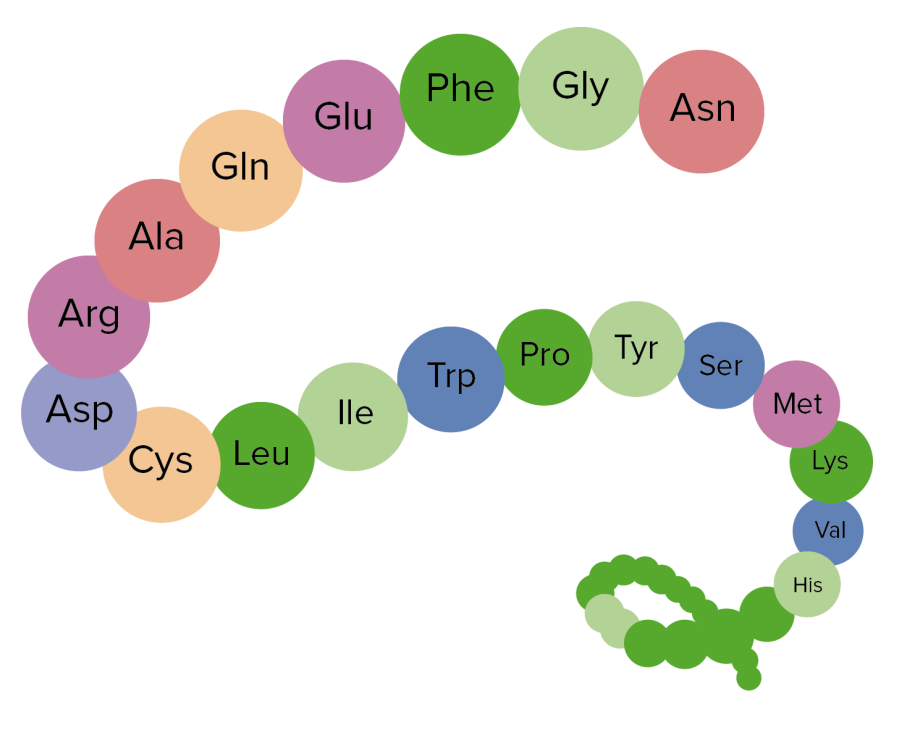
Na imagem apresentada é possível visualizar a estrutura primária das proteínas (uma agregação de aminoácidos)
Imagem por Lecturio.Estrutura secundária:
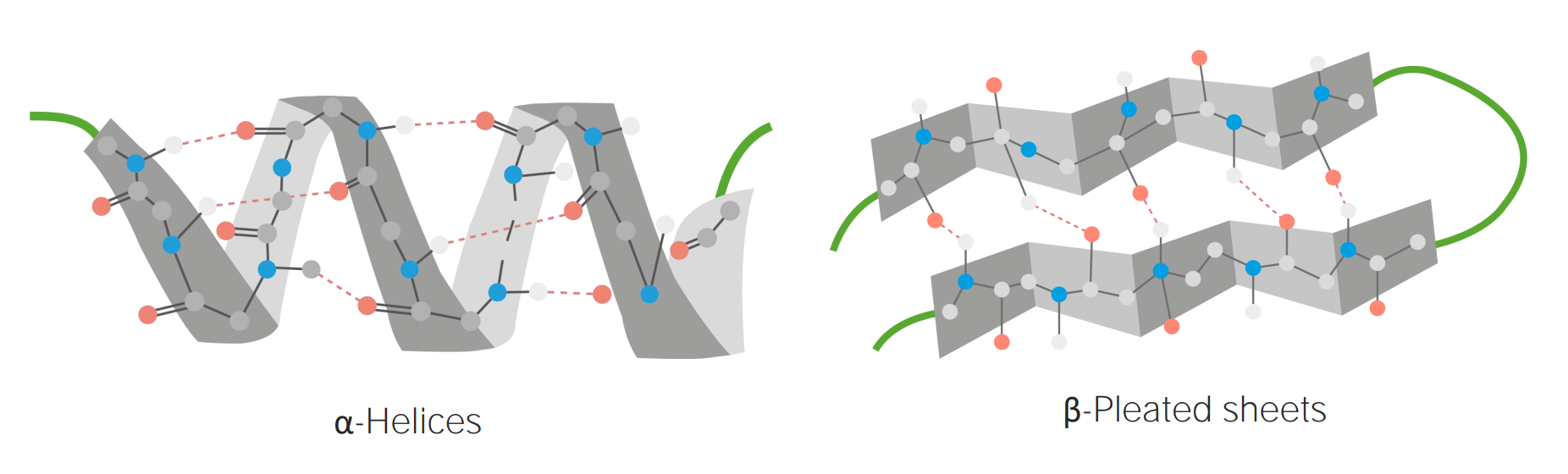
Exemplos de estuturas em α-hélices e folhas β-plissadas
Imagem por Lecturio.Estrutura terciária:

Exemplo de uma estrutura terciária
Imagem por Lecturio.
A imagem apresentada representa a estrutura terciária das proteínas
Imagem por Lecturio.Estrutura quaternária:
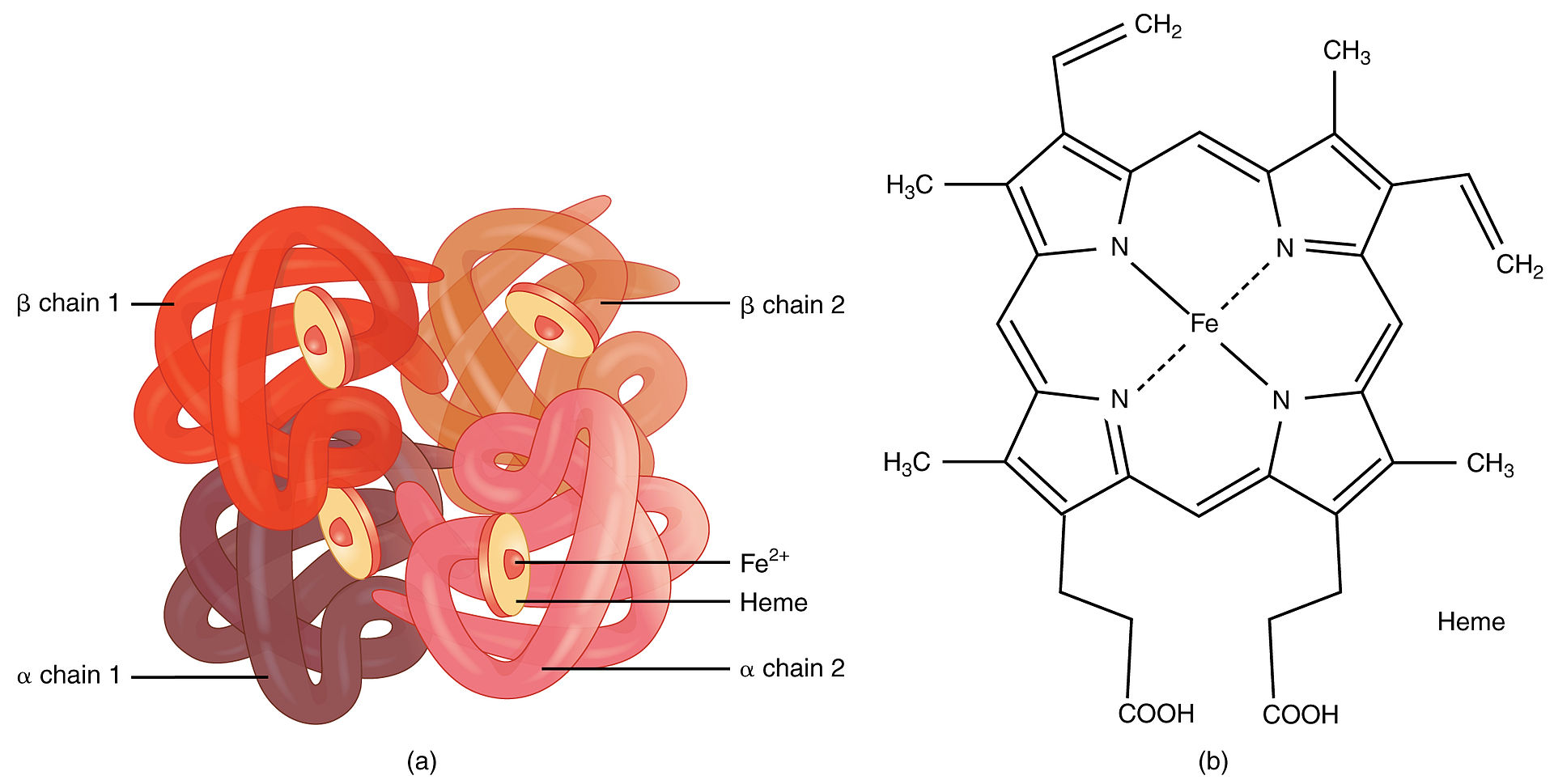
Hemoglobina:
Um exemplo de estrutura quaternária
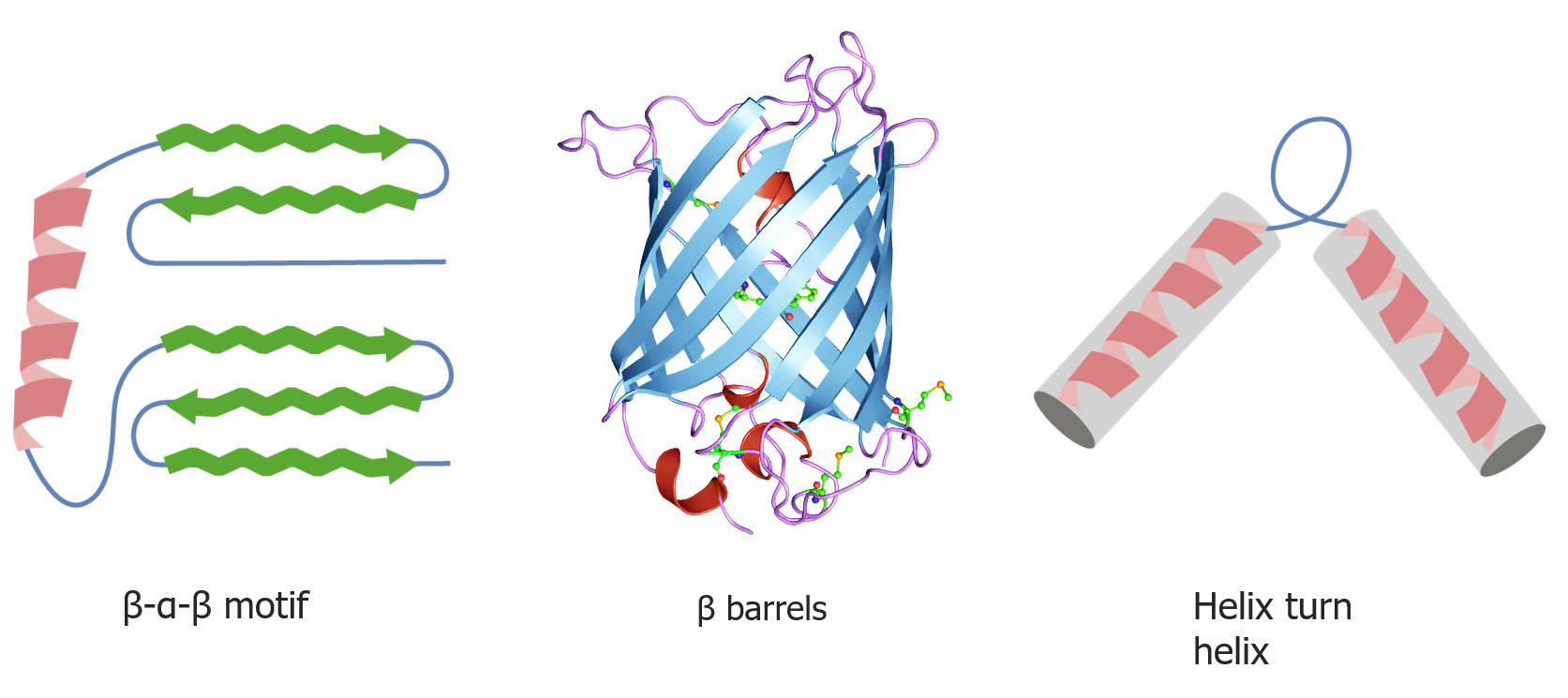
Formas do “folding” de proteínas quaternárias e terciárias
Imagem por Lecturio.As proteínas “chaperones” ajudam no “folding” das proteínas.
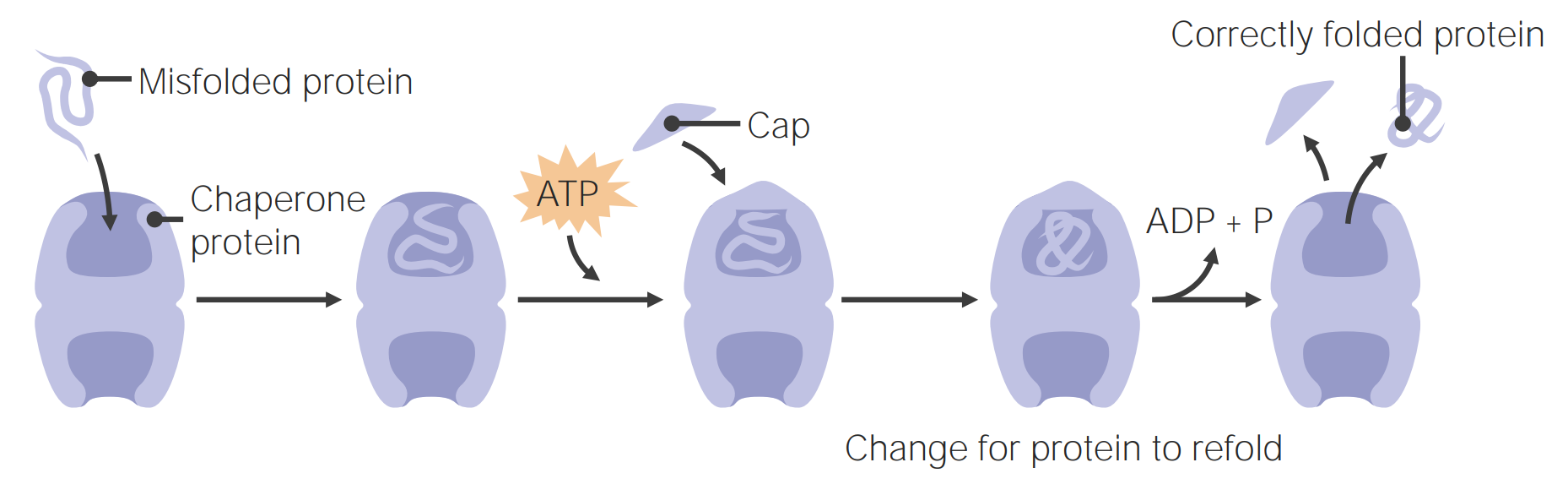
As proteínas “chaperones” auxiliam no “folding” das proteínas
Imagem por Lecturio.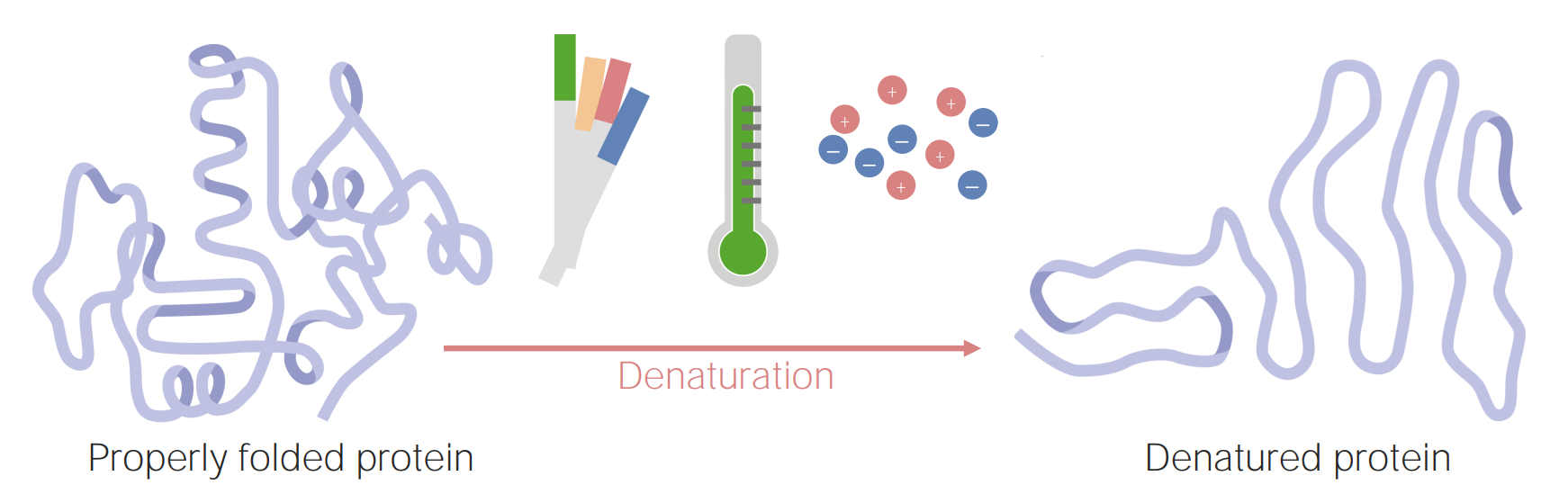
As proteínas podem ficar desnaturadas (ou mal estruturadas) como resultado de alterações no pH, temperatura ou concentração iónica.
Imagem por Lecturio.A estrutura única da proteína (primária, secundária, terciária e quaternária) confere à mesma, propriedades físicas e químicas importantes para a sua função. Algumas dessas propriedades são:
As proteínas apresentam uma grande variedade de funções no corpo, incluindo:
| Enzima | Zimógeno (precursor) | Ativado por | Notas sobre a atividade |
|---|---|---|---|
| Enzimas gástricas secretadas no estômago | |||
| Pepsina | Pepsinogénio | Ácido clorídrico | Mais MAIS Androgen Insensitivity Syndrome eficiente entre AAs hidrofóbicos |
| Enzimas pancreáticas secretadas no duodeno | |||
| Tripsina | Tripsinogénio | Enteropeptidase Enteropeptidase A specialized proteolytic enzyme secreted by intestinal cells. It converts trypsinogen into its active form trypsin by removing the n-terminal peptide. Digestion and Absorption |
|
| Quimotripsina | Quimotripsinogénio | Tripsina | Mais MAIS Androgen Insensitivity Syndrome eficiente entre AAs hidrofóbicos |
| Carboxipeptidase | Procarboxipeptidase | Tripsina |
|
| Elastase Elastase A protease of broad specificity, obtained from dried pancreas. Molecular weight is approximately 25, 000. The enzyme breaks down elastin, the specific protein of elastic fibers, and digests other proteins such as fibrin, hemoglobin, and albumin. Proteins and Peptides | Proelastase | Tripsina | Igual à atividade da carboxipeptidase |
| Enzimas ligadas à borda em escova dos enterócitos no intestino delgado | |||
| Aminopeptidase Aminopeptidase A subclass of exopeptidases that act on the free n terminus end of a polypeptide liberating a single amino acid residue. Digestion and Absorption | N / D | N / D | Quebra a ligação dos pequenos peptídeos do terminal amino (ou seja, do N-terminal) |
| Dipeptidases Dipeptidases Exopeptidases that specifically act on dipeptides. Proteins and Peptides | N / D | N / D | Quebra as ligações peptídicas entre 2 AAs → 2 AAs simples |
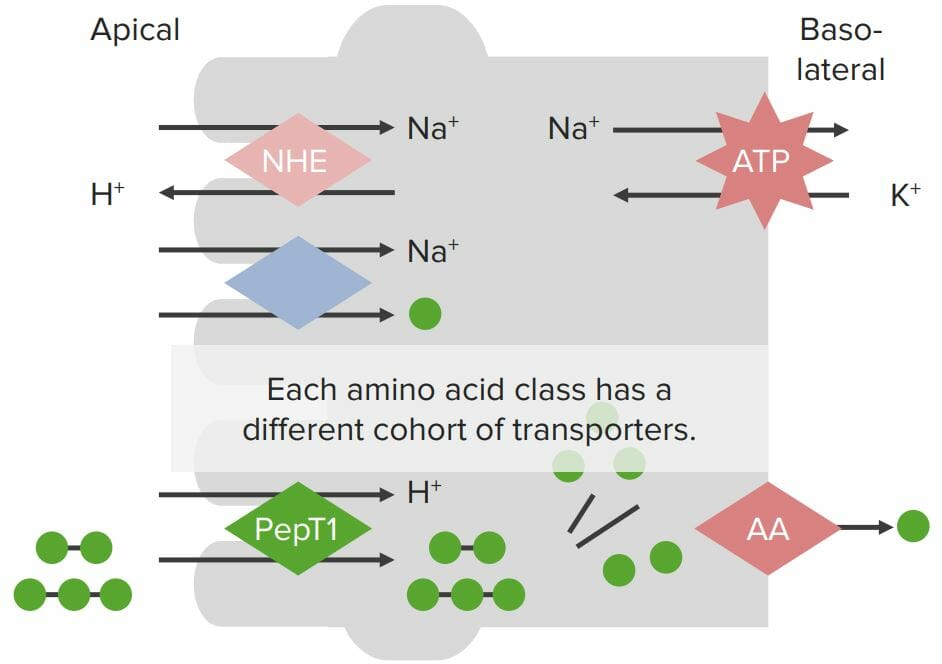
Proteínas de transporte presentes nas membranas dos enterócitos e envolvidas na absorção de proteínas:
A ATPase Na+/K + presente na membrana basolateral cria um gradiente de Na + no interior da célula. O transportador Na +/H + (NHE, pela sigla em inglês) na membrana apical também é responsável pela criação de um gradiente H +. Os aminoácidos individuais (AAs; bolas verdes) são absorvidos através de um cotransportador Na +/AA, onde o Na + se desloca através da membrana apical para os enterócitos a favor do gradiente de concentração, movimentando o AA com ele (apesar de se deslocar contra o gradiente químico de AA). Os pequenos peptídeos são absorvidos através do cotransportador H+/PepT juntamente com o H +, deslocando-se a favor do gradiente de concentração para dentro da célula, transportando os pequenos peptídeos com ele. Dentro dos enterócitos, os pequenos peptídeos são decompostos em AAs individuais por ação das peptidases. Todos os AAs são então absorvidos via transportadores específicos presentes na membrana basolateral.
O metabolismo proteico consiste num grupo de processos bioquímicos responsável tanto pelo anabolismo (síntese de proteínas e AAs) como pelo catabolismo (decomposição de proteínas e AAs).
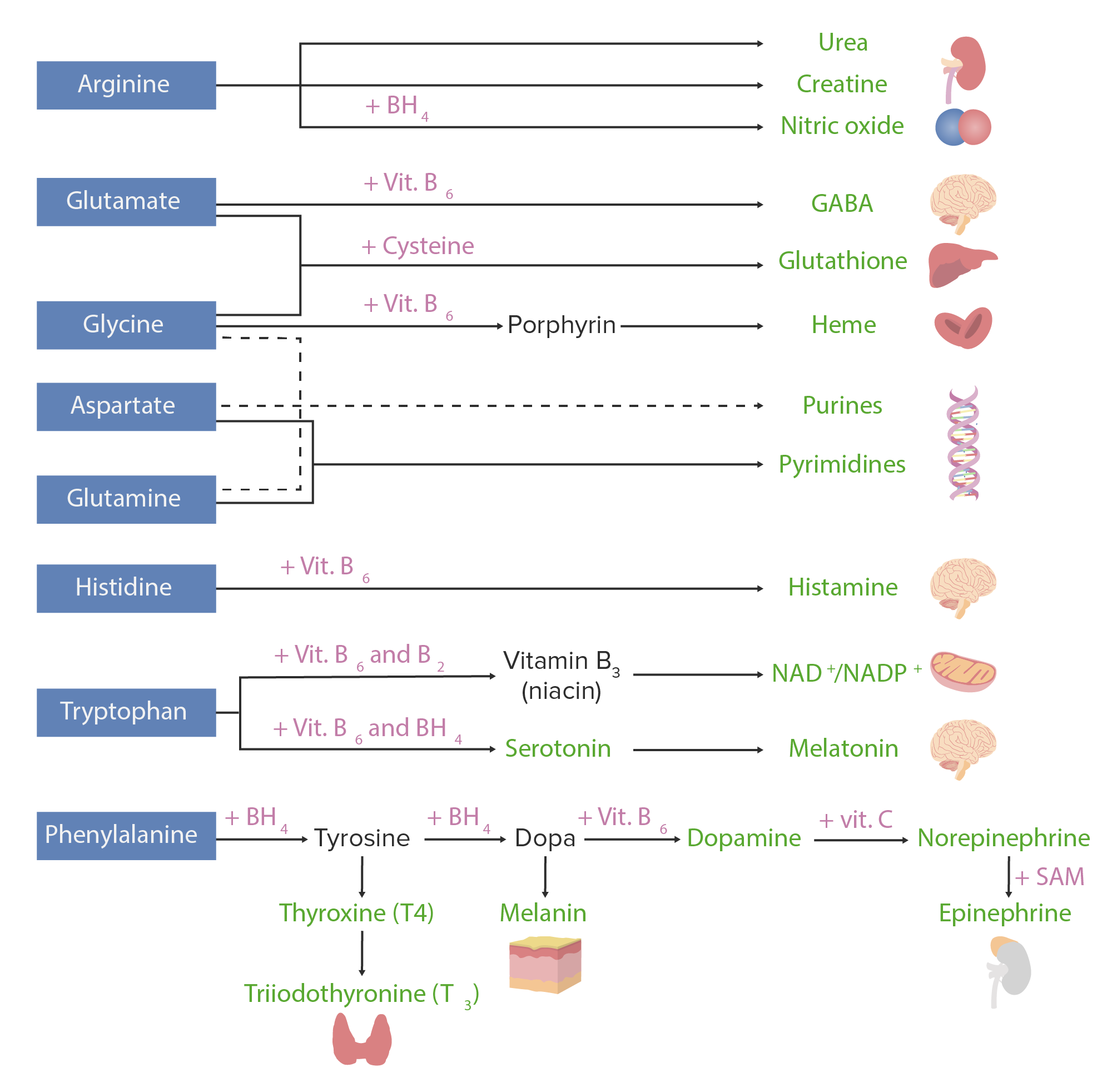
Derivados dos aminoácidos:
Os aminoácidos (com cor azul) associam-se a determinados cofatores ou a outros substratos (com cor rosa) para dar origem a várias substâncias biologicamente importantes (representadas a verde).
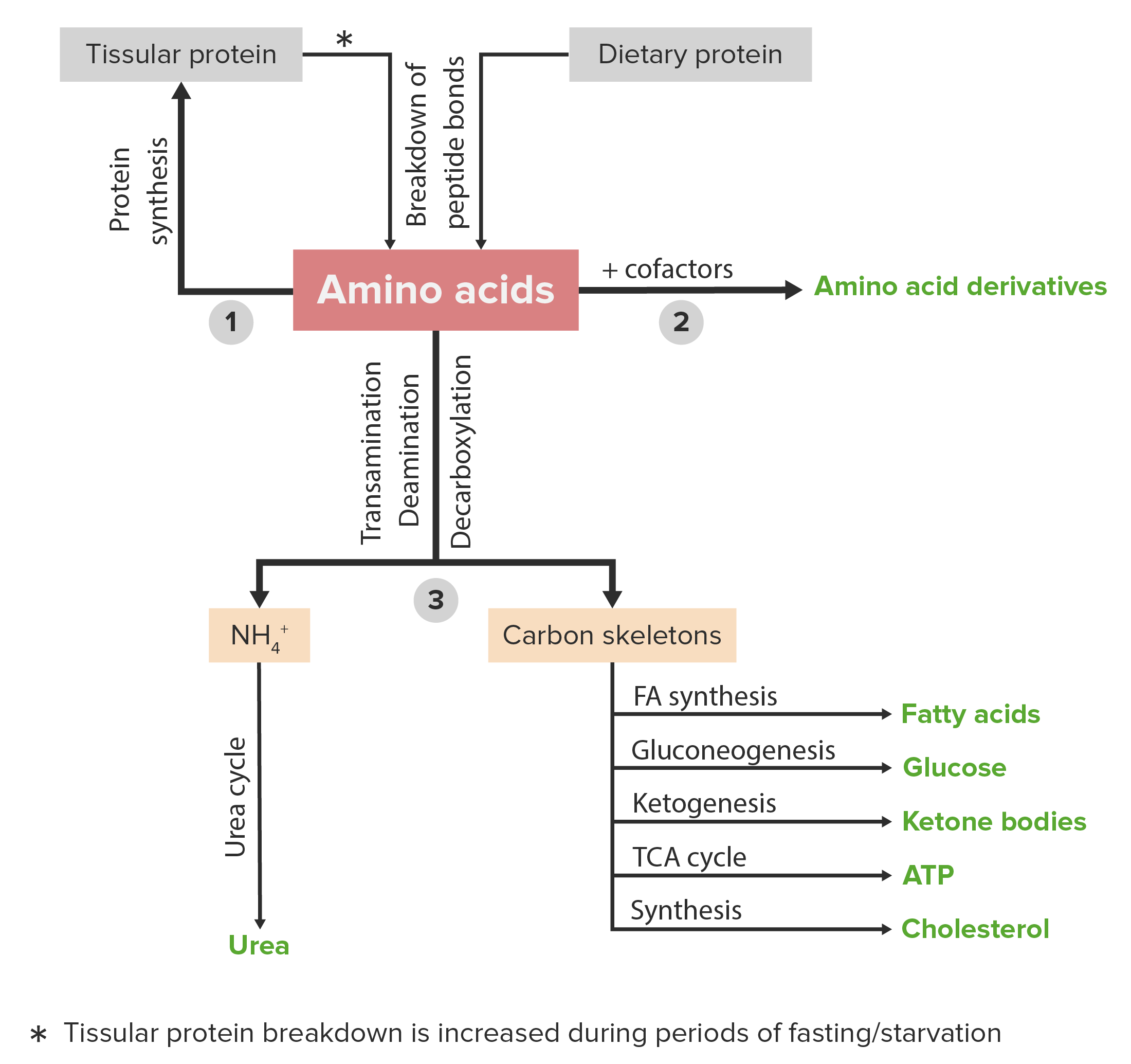
A imagem apresentada representa esquemáticamente o metabolismo dos aminoácidos, incluindo as 3 principais vias: reutilização na síntese de novas proteínas, associação com cofatores para a produção de derivados de aminoácidos e o catabolismo. O catabolismo dos aminoácidos inclui a remoção de grupos funcionais e a decomposição do esqueleto de carbono.
Imagem por Lecturio.As anomalias ou défices de proteínas e/ou anomalias relacionadas com o seu metabolismo são responsáveis por inúmeras patologias. Alguns exemplos das mesmas encontram-se listados abaixo.
