


A glicólise é uma via metabólica central responsável pela quebra de glucose Glucose A primary source of energy for living organisms. It is naturally occurring and is found in fruits and other parts of plants in its free state. It is used therapeutically in fluid and nutrient replacement. Lactose Intolerance e tem um papel vital na geração de energia livre para a célula e metabolitos para posterior degradação oxidativa. A glucose Glucose A primary source of energy for living organisms. It is naturally occurring and is found in fruits and other parts of plants in its free state. It is used therapeutically in fluid and nutrient replacement. Lactose Intolerance fica disponível principalmente no sangue como resultado da degradação do glicogénio ou da sua síntese a partir de precursores não carboidratos (gluconeogénese), sendo importada para as células por proteínas transportadoras específicas. A glicólise ocorre no citoplasma e consiste em 10 reacções, cujo resultado líquido é a conversão de 1 glucose Glucose A primary source of energy for living organisms. It is naturally occurring and is found in fruits and other parts of plants in its free state. It is used therapeutically in fluid and nutrient replacement. Lactose Intolerance C6 em 2 moléculas de piruvato C3. A energia livre deste processo é recolhida para produzir adenosina trifosfato (ATP) e nicotinamida adenina dinucleótido hidreto (NADH), metabolitos-chave para a produção de energia. A estequiometria geral do percurso é: glucose Glucose A primary source of energy for living organisms. It is naturally occurring and is found in fruits and other parts of plants in its free state. It is used therapeutically in fluid and nutrient replacement. Lactose Intolerance + 2 Pi + 2 ADP + 2 NAD NAD+ A coenzyme composed of ribosylnicotinamide 5'-diphosphate coupled to adenosine 5'-phosphate by pyrophosphate linkage. It is found widely in nature and is involved in numerous enzymatic reactions in which it serves as an electron carrier by being alternately oxidized (NAD+) and reduced (NADH). Pentose Phosphate Pathway+> 2 piruvato + 2 ATP + 2 NADH + 2 H+ + 2 H2O (H+: ião hidrogénio, Pi: ião fosfato, NAD NAD+ A coenzyme composed of ribosylnicotinamide 5'-diphosphate coupled to adenosine 5'-phosphate by pyrophosphate linkage. It is found widely in nature and is involved in numerous enzymatic reactions in which it serves as an electron carrier by being alternately oxidized (NAD+) and reduced (NADH). Pentose Phosphate Pathway+: nicotinamida adenina dinucleótido).
Last updated: Apr 25, 2025
A 1.ª metade da glicólise requer um investimento energético de 2 moléculas de adenosina trifosfato (ATP) e serve para converter a glucose Glucose A primary source of energy for living organisms. It is naturally occurring and is found in fruits and other parts of plants in its free state. It is used therapeutically in fluid and nutrient replacement. Lactose Intolerance hexagonal em 2 trioses. O processo consiste em 5 etapas:
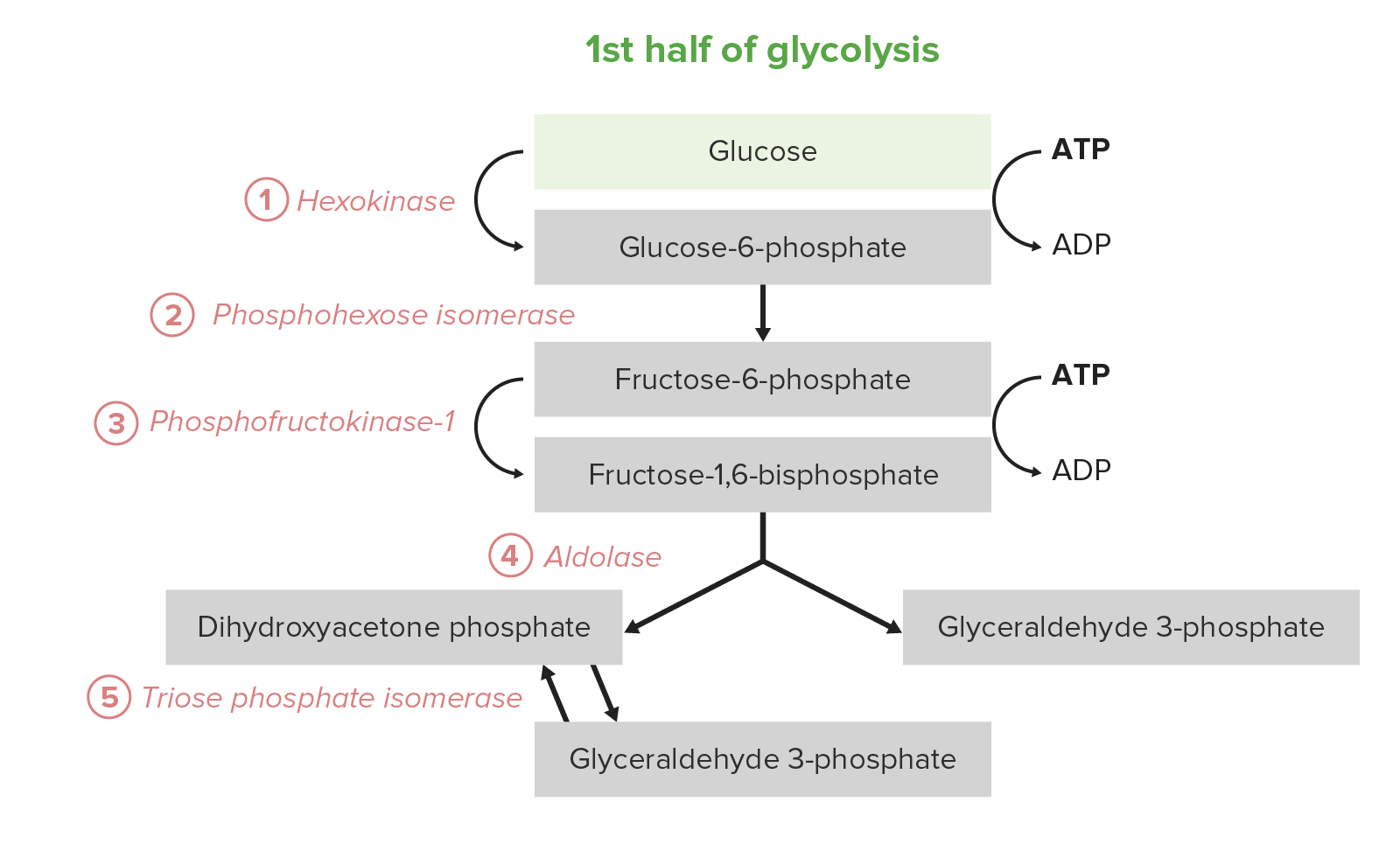
Os 5 primeiros passos (primeira metade) do percurso da glicólise
Image by Lecturio.A segunda metade da glicólise converte a triose GAP em piruvato, com a geração concomitante de 4 ATP e 2 nicotinamida adenina dinucleótido hidreto (NADH) por cada 2 GAP. Assim, o investimento energético dos passos 1–5 é pago duas vezes aqui. Em certos tipos de células e condições, estes 5 passos são a fonte predominante de ATP:
Reacção líquida: glucose Glucose A primary source of energy for living organisms. It is naturally occurring and is found in fruits and other parts of plants in its free state. It is used therapeutically in fluid and nutrient replacement. Lactose Intolerance + 2 Pi + 2 ADP + 2 NAD NAD+ A coenzyme composed of ribosylnicotinamide 5′-diphosphate coupled to adenosine 5′-phosphate by pyrophosphate linkage. It is found widely in nature and is involved in numerous enzymatic reactions in which it serves as an electron carrier by being alternately oxidized (NAD+) and reduced (NADH). Pentose Phosphate Pathway+ → 2 piruvato + 2 ATP + 2 NADH + 2 H+ + 2 H2O
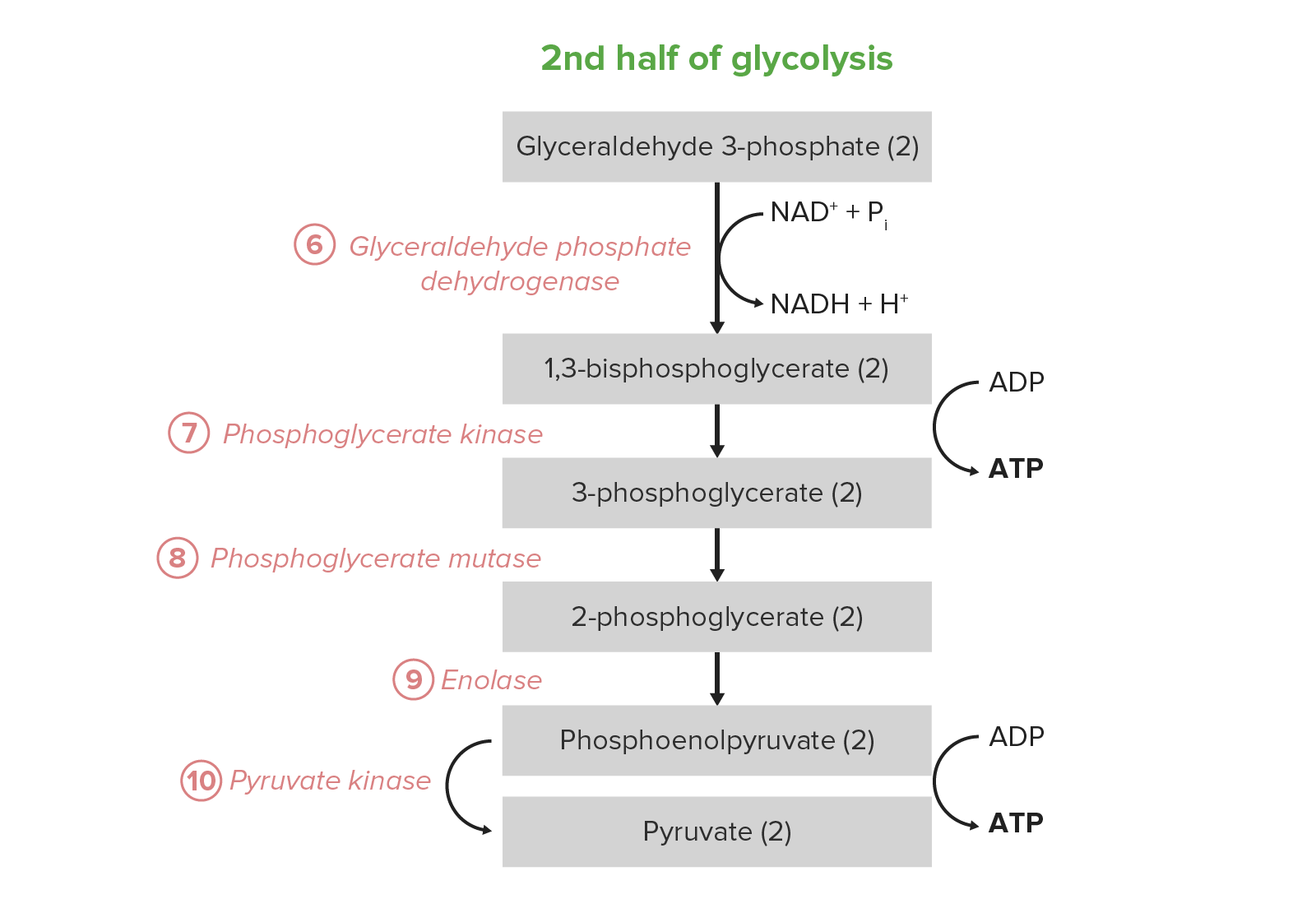
Os últimos 5 passos (última metade) da via da glicólise.
Image by Lecturio.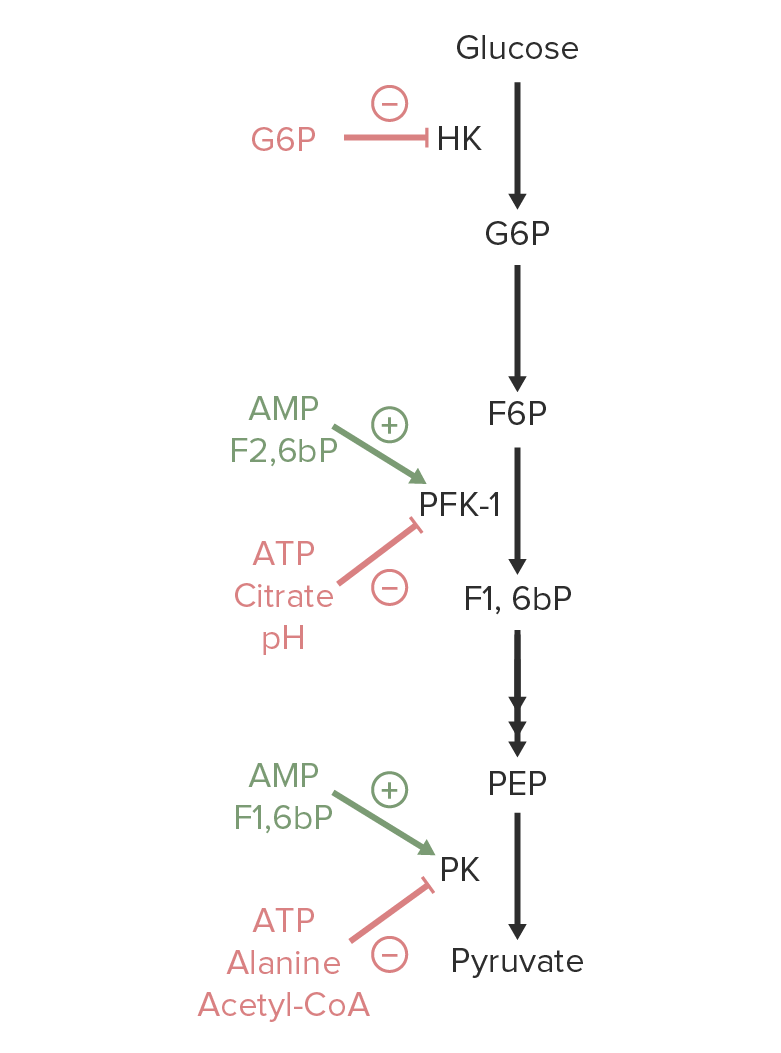
Uma visão geral da regulação da glicólise. Os ativadores da hexocinase (HK), da fosfofrutocinase-1 (PFK-1), ou a piruvato cinase (PK) estão marcados a verde. Os metabolitos que inibem estas enzimas estão marcados a vermelho.
Image by Lecturio.De seguida estão apresentadas enzimas da via da glicólise que podem estar envolvidas em defeitos enzimáticos congénitos:
Estes defeitos enzimáticos congénitos produzem anemia Anemia Anemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types hemolítica.
Anemia Anemia Anemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types hemolítica: grupo de anemias que se devem à destruição ou eliminação prematura de hemácias. Anomalias intrínsecas de hemácias levam à depuração esplénica (hemólise extravascular). A destruição crónica de hemácias pode-se apresentar como icterícia, esplenomegalia, colelitíase, hematúria e sintomas de anemia Anemia Anemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types (falta de ar AR Aortic regurgitation (AR) is a cardiac condition characterized by the backflow of blood from the aorta to the left ventricle during diastole. Aortic regurgitation is associated with an abnormal aortic valve and/or aortic root stemming from multiple causes, commonly rheumatic heart disease as well as congenital and degenerative valvular disorders. Aortic Regurgitation, fadiga, síncope e taquicardia).
