


Os aminoácidos são os blocos de construção das proteínas, cuja produção é uma cascata enzimática regulada de forma rigorosa. As doenças das enzimas envolvidas no metabolismo de aminoácidos são muitas vezes graves e apresentam-se no início da vida. Os erros congénitos no metabolismo de aminoácidos devem-se a uma síntese ou degradação deficiente. Nos Estados Unidos, os recém-nascidos são rotineiramente examinados no nascimento a doenças comuns no metabolismo de aminoácidos, que incluem fenilcetonúria (PKU, pela sigla em inglês), leucinose, homocistinúria, tirosinemia e alcaptonúria. Os sintomas destas doenças apresentam-se frequentemente no início da vida. Embora o tratamento varie, a maioria destas doenças requer mudanças dietéticas e algumas requerem suplementação proteica ou outros regimes de medicação.
Last updated: May 19, 2025
A fenilcetonúria (PKU, pela sigla em inglês) é uma doença hereditária que provoca um aumento dos níveis de fenilalanina no organismo devido à incapacidade de metabolizar este aminoácido.
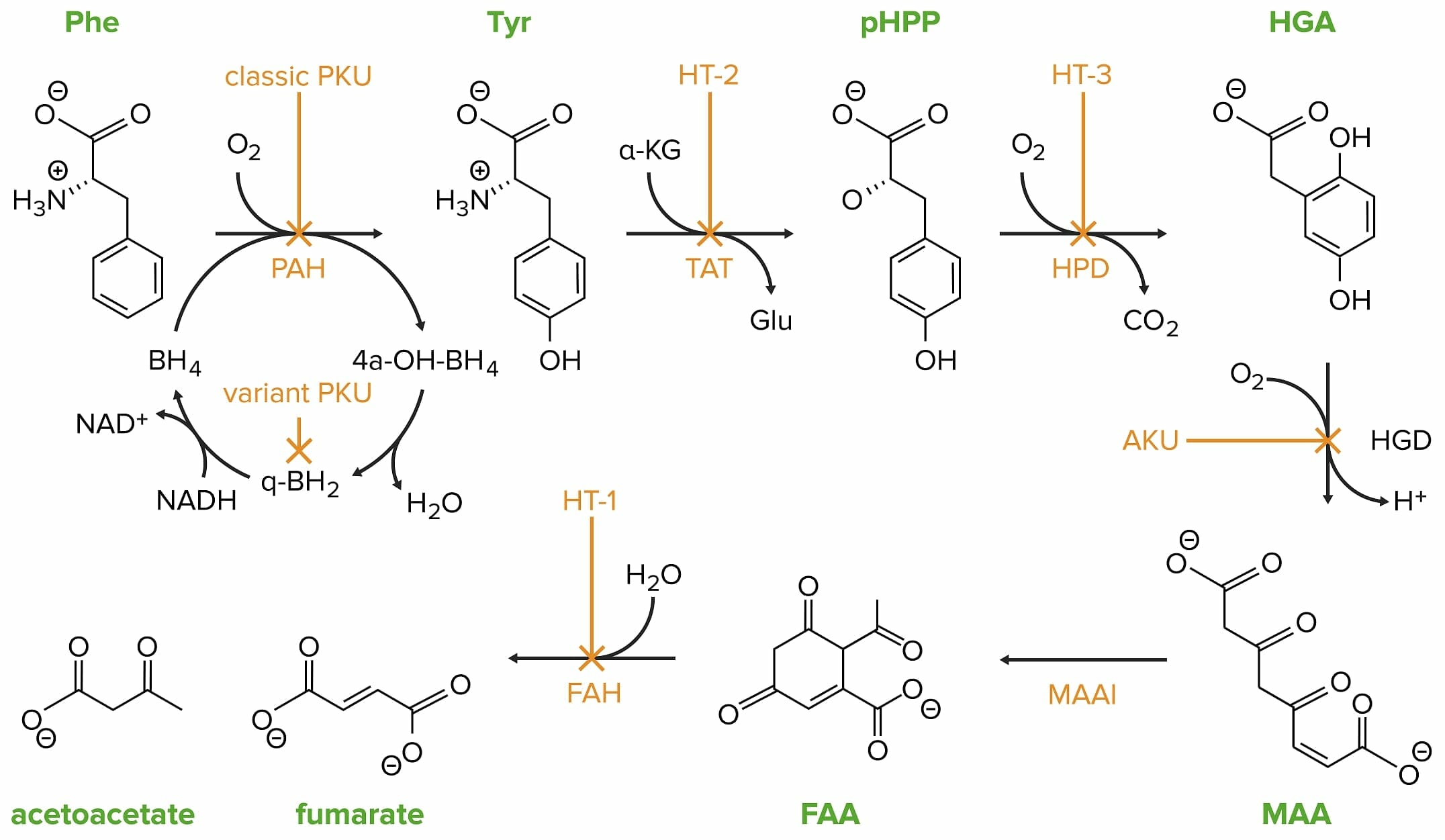
A fenilcetonúria clássica (PKU) é causada por uma mutação no gene da fenilalanina hidroxilase (PAH).
Phe: fenilalanina
PAH: fenilalanina hidroxilase
Tyr: tirosina
HT-2: tirosinemia II
TAT: tirosina transaminase
pHPP: p-hidroxifenilpiruvato
HPD: p-hidroxifenilpiruvato desoxigenase
HT-3: tirosinemia III
HGA: ácido homogentísico
HGD: ácido homogentísico oxidase
AKU: alcaptonuria
MAA: 4-maleilacetoacetato
MAAI: 4-maleilacetoacetato isomerase
FAA: 4-fumarilacetoacetato
FAH: 4-fumarilacetoacetase
HT-1: tirosinemia I
O sangue de um bebé de 2 semanas é recolhido para um rastreio de fenilcetonúria (PKU).
Imagem: “Phenylketonuria testing” by USAF Photographic Archives. Licença: Public DomainEsta doença não tem cura e deve ser tratada com modificação na alimentação e medicação/suplementação.
A cistinúria é uma causa genética de cálculos renais devido à limitação no transporte renal de cistina. Há diminuição da reabsorção tubular proximal da cistina filtrada resultando no aumento da excreção urinária de cistina e na formação de cálculos de cistina.
Suspeita clínica:
Testes Testes Gonadal Hormones confirmatórios:
A cistinúria é uma doença metabólica hereditária do metabolismo da metionina que resulta na elevação dos níveis plasmáticos e urinários de homocisteína e causa uma gama de manifestações clínicas.
Há duas vias metabólicas normais da metionina:
A elevações dos níveis plasmáticos de homocisteína resultam primariamente de:
Outras causas de elevação plasmática de homocisteína:
Suspeita clínica:
Testes Testes Gonadal Hormones confirmatórios:
Uma doença genética que se manifesta quando há alterações do metabolismo da tirosina, um aminoácido envolvido na síntese das hormonas tiroideias, das catecolaminas e da melanina.
Em condições não patológicas, a tirosina é metabolizada pelos hepatócitos e pelos túbulos renais proximais em acetoacetato (cetogénico) e fumarato (glucogénico).
Defeitos genéticos:
HT1:
HT2:
HT3:
AKU:
Suspeita clínica:
Testes Testes Gonadal Hormones confirmatórios:
Um grupo de doenças genéticas do metabolismo de aminoácidos envolvendo aminoácidos de cadeia ramificada como leucina, isoleucina e valina. Estas doenças são causados pela perturbação do metabolismo destes aminoácidos, resultando no acúmulo de metabolitos tóxicos que muitas vezes se espalham na urina.
Existem 4 tipos principais de acidemia Acidemia Respiratory Acidosis orgânica:
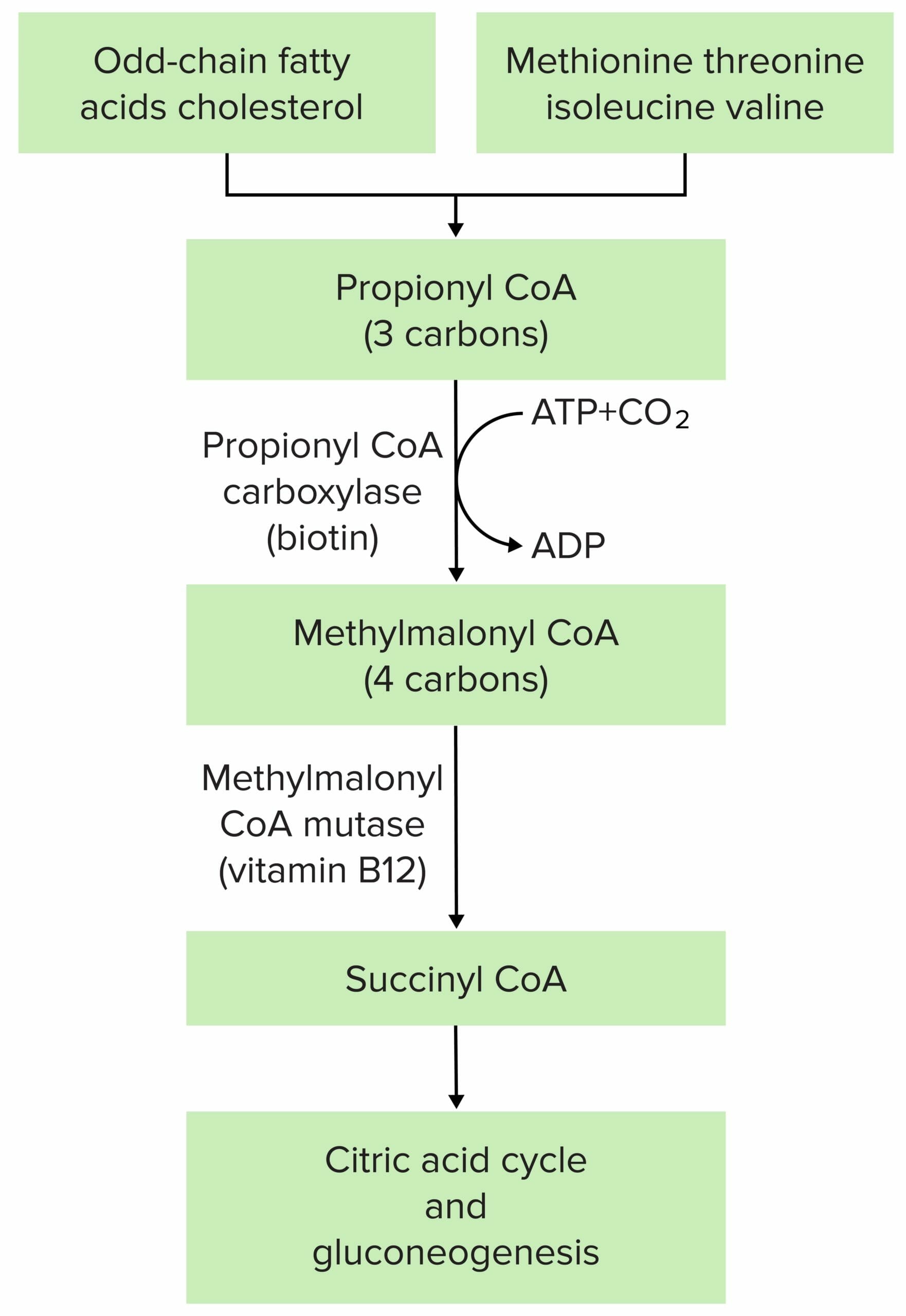
A fisiopatologia da acidemia metilmalónica é mais frequentemente devida à mutação da metilmalonil-CoA mutase ou da metilmalonil-CoA epimerase.
Imagem por Lecturio.