


As doenças do armazenamento do glicogénio (GSDs, pela sigla em inglês) consistem em defeitos genéticos que levam a perturbações no metabolismo dos carbohidratos. Estas doenças são causadas pela presença de variantes genéticas patogénicas que afetam as enzimas envolvidas na degradação do glicogénio. O défice de uma dessas enzimas pode ocorrer no fígado ou nos músculos e pode causar hipoglicemia e/ou deposição anormal de glicogénio nos tecidos. As apresentações clínicas são variáveis, desde uma apresentação fatal no período neonatal até à apresentação inicial com sintomas na idade adulta. Existem pelo menos 14 formas de GSDs, sendo as 4 mais MAIS Androgen Insensitivity Syndrome frequentes e importantes, a doença de Von Gierke, a doença de Pompe, a doença de Cori e a doença de McArdle. O diagnóstico é clínico; a biópsia permite detetar a presença de glicogénio nos tecidos e a doença é confirmada através da análise do ADN. O tratamento visa tratar ou evitar episódios de hipoglicemia, hiperuricemia, dislipidemia (HLD, pela sigla em inglês) e acidose láctica. Atualmente, não existe nenhum tratamento curativo, mas estão a ser experimentados tratamentos genéticos
Last updated: Apr 17, 2025
As doenças do armazenamento de glicogénio (GSDs, pela sigla em inglês) são caracterizadas pela presença de defeitos genéticos que causam défices enzimáticos, que resultam na deposição anormal de glicogénio nos tecidos, com o consequente aparecimento de doenças hepáticas, musculares ou cardíacas, e episódios de hipoglicemia (na maioria das patologias), uma vez que o corpo não pode utilizar o glicogénio como fonte de energia.
A doença de armazenamento do glicogénio I (doença de von Gierke) é causada por mutações que levam a défices enzimáticos; estes, por sua vez, resultam na acumulação excessiva de glicogénio e de gordura nos tecidos, e em episódios de hipoglicemia.
A doença de armazenamento do glicogénio II (doença de Pompe) corresponde a uma patologia de armazenamento lisossómico autossómica recessiva, que é causada pela presença de uma variante patogénica no GAA. Este defeito resulta no défice da enzima alfa-glicosidase, com a subsequente acumulação de glicogénio no músculo cardíaco e esquelético.
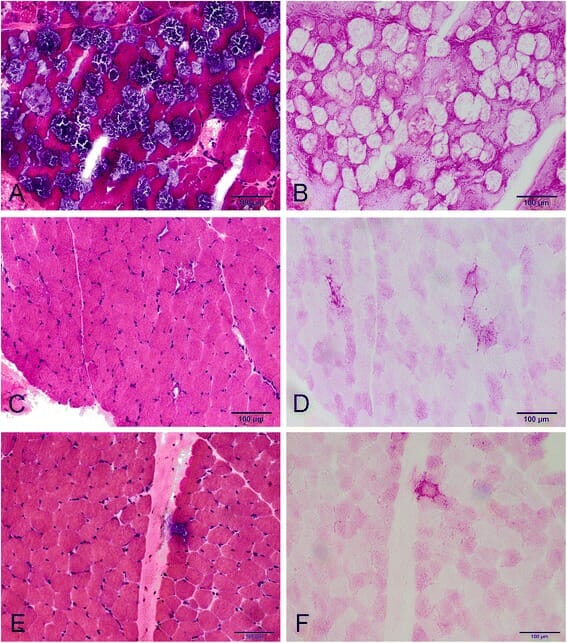
Alterações miopáticas na doença de Pompe com início tardio:
A: Coloração H&E – presença de vacuolização extensa em muitas fibras.
C e E: coloração H&E – presença da vacuolização apenas algumas fibras, num doente diferente.
B, D e F: coloração PAS – fibras vacuolares com o glicogénio a corar positivamente.
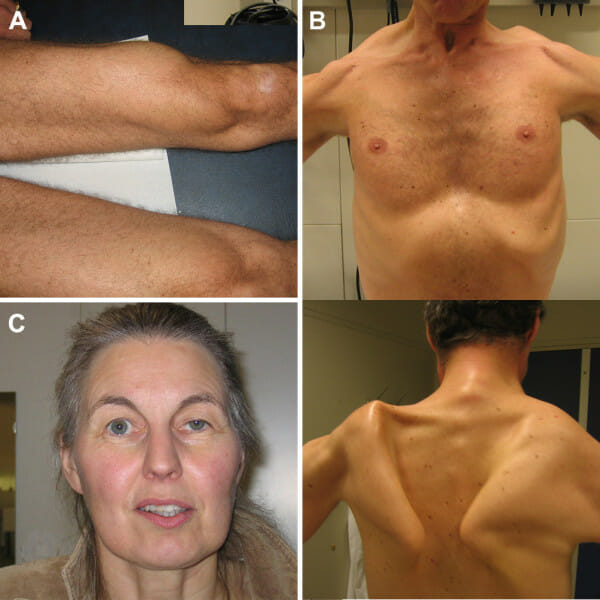
Características clínicas na doença de Pompe:
A atrofia do músculo quadríceps (A), escapula alada (B) e a ptose (C) são as principais características clínicas evidentes em adultos com doença de Pompe. As fotografias foram expostas com o permissão dos doentes.
A doença de armazenamento do glicogénio III (doença de Cori) é causada pelo défice da enzima de desramificação do glicogénio, com consequente deposição de glicogénio no fígado, músculo e coração.
A doença de armazenamento do glicogénio V (doença de McArdle), também conhecida como défice de miofosforilase, é uma patologia autossómica recessiva causada por mutações na isoforma muscular da fosforilase (glicogénio fosforilase muscular, ou PYGM) localizada no 11q13.
| von Gierke ( GSD I GSD I An autosomal recessive disease in which gene expression of glucose-6-phosphatase is absent, resulting in hypoglycemia due to lack of glucose production. Accumulation of glycogen in liver and kidney leads to organomegaly, particularly massive hepatomegaly. Increased concentrations of lactic acid and hyperlipidemia appear in the plasma. Clinical gout often appears in early childhood. Glycogen Storage Disorders) | Pompe ( GSD II GSD II An autosomal recessively inherited glycogen storage disease caused by glucan 1, 4-alpha-glucosidase deficiency. Large amounts of glycogen accumulate in the lysosomes of skeletal muscle; heart; liver; spinal cord; and brain. Three forms have been described: infantile, childhood, and adult. The infantile form is fatal in infancy and presents with hypotonia and a hypertrophic cardiomyopathy. The childhood form usually presents in the second year of life with proximal weakness and respiratory symptoms. The adult form consists of a slowly progressive proximal myopathy. Glycogen Storage Disorders) | Cori ( GSD III GSD III An autosomal recessive metabolic disorder due to deficient expression of amylo-1, 6-glucosidase (one part of the glycogen debranching enzyme system). The clinical course of the disease is similar to that of glycogen storage disease type I, but milder. Massive hepatomegaly, which is present in young children, diminishes and occasionally disappears with age. Levels of glycogen with short outer branches are elevated in muscle, liver, and erythrocytes. Six subgroups have been identified, with subgroups type IIIa and type IIIb being the most prevalent. Glycogen Storage Disorders) | McArdle ( GSD V GSD V Glycogenosis due to muscle phosphorylase deficiency. Characterized by painful cramps following sustained exercise. Glycogen Storage Disorders) | |
|---|---|---|---|---|
| Apresentação clínica |
|
|
|
|
| Diagnóstico |
|
|
|
|
| Tratamento |
|
|
|
|
