


A doença de Niemann-Pick (DNP), também conhecida como deficiência de esfingomielinase, é uma doença de armazenamento lisossomal hereditário raro. A doença é classificada com base na mutação genética. O tipo A e o tipo B derivam de mutações no gene SMPD-1, resultando na deficiência da enzima esfingomielinase ácida. O tipo C resulta de mutações nos genes NPC1 ou NPC2 , que são necessárias para o transporte intracelular de lípidos. Estas mutações resultam em lisossomas incapazes de metabolizar adequadamente a gordura, como a esfingomielina e o colesterol, o que resulta na acumulação progressiva de lípidos intracelulares e danos nos órgãos. As manifestações clínicas podem incluir má progressão estaturo-ponderal, hepatoesplenomegalia, trombocitopenia, doença pulmonar intersticial, comprometimento cognitivo e motor, e manchas maculares vermelho-cereja ("cherry-red spots"). São observadas neurodegeneração progressiva e uma esperança média de vida curta no DNP-A, enquanto o DNP-B é tipicamente não neurogénico. O diagnóstico de DNP é baseado na suspeita clínica e pode ser confirmado pela medição da atividade da esfingomielinase ou biomarcadores, teste genético ou biópsia. Atualmente, não há cura para o DNP, portanto, o tratamento é de suporte e concentra-se no controlo dos sintomas.
Last updated: Dec 15, 2025
A doença de Niemann-Pick (DNP) é uma doença de armazenamento lisossómico autossómica recessiva, classificada com base na mutação genética e na deficiência enzimática:
Lisossomas normais:
Em NPD-A e B:
Em NPD-C:
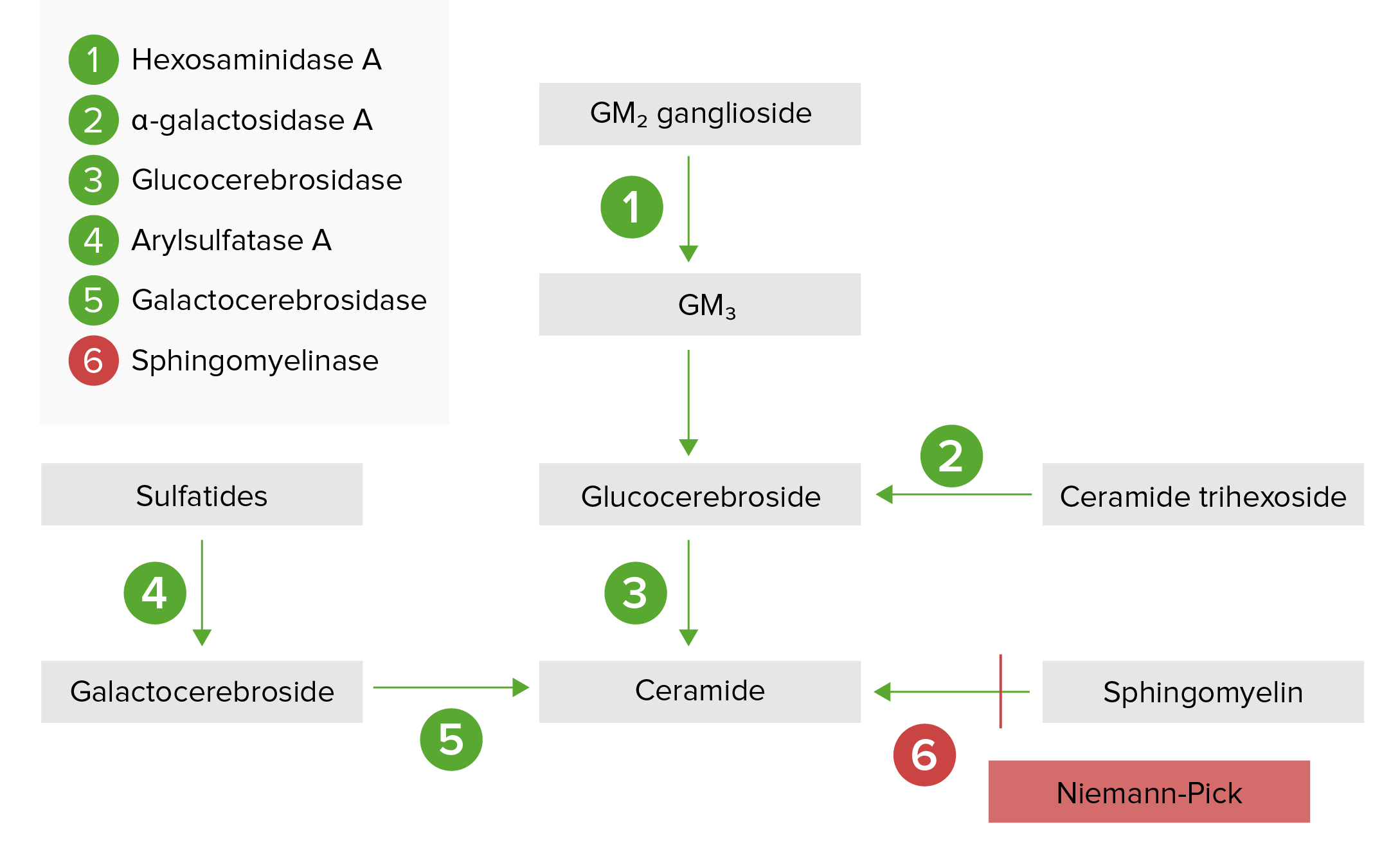
A via de armazenamento lisossomal:
A doença de Niemann-Pick (tipos A e B) resulta da deficiência de esfingomielinase ácida, que resulta na acumulação de esfingomielina.
A apresentação clínica da DNP depende do tipo e gravidade da doença.
Curso da doença:
Sinais e sintomas:
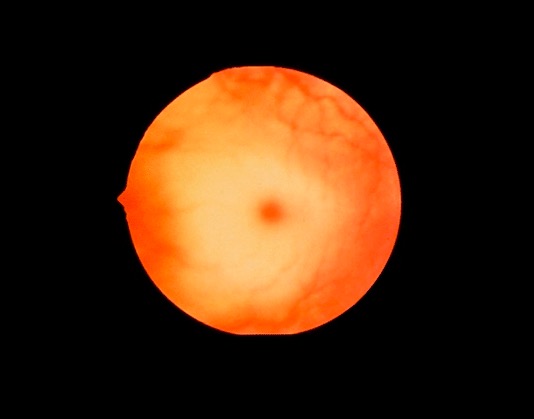
Imagem fundoscópica que mostra uma “cherry-red spot”, vista na doença de Niemann-Pick
Imagem : “Cherry red spot” por Jonathan Trobe, MD Licença: CC BY 3.0Curso da doença:
Sinais e sintomas:
Curso da doença:
Sinais e sintomas:
A suspeita de DNP é levantada com base nas características clínicas. O diagnóstico pode ser confirmado com as seguintes investigações:
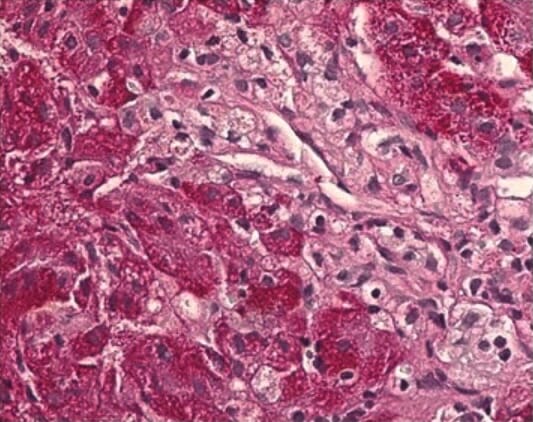
Biópsia histopatológica do fígado que mostra células de Kupffer aumentadas com um citoplasma espumoso, típico da DNP-C
Imagem: “Histopathological liver biopsy findings” por Federal State Budget Institution, Research Center for Obstetrics, Gynecology and Perinatology, Federal State Budget Institution, 117997 Oparina str. 4, Moscow, Russia. Licença: CC BY 4.0Não há cura para a DNP. O tratamento é de suporte e multidisciplinar.
