


A doença de Hirschsprung (DH), também conhecida como aganglionose congénita ou megacólon congénito, é uma anomalia congénita do cólon causada pela falha na migração das células ganglionares derivadas da crista neural até ao cólon distal. A ausência de inervação envolve sempre o reto e estende-se proximalmente e de forma contigua até distâncias variáveis. Geralmente, o diagnóstico é feito no período neonatal, através da manifestação da tríade clássica de sintomas, que inclui o atraso na passagem de mecônio, a distensão abdominal e os vómitos biliares. As crianças que apresentam graus mais MAIS Androgen Insensitivity Syndrome ligeiros de obstrução funcional podem não ser diagnosticadas até à infância tardia, altura em que começam a manifestar obstipação crónica refratária, distensão abdominal e défice de crescimento. O diagnóstico de DH é confirmado pela ausência de células ganglionares na biópsia retal após a realização de exames não invasivos, como a manometria anorretal e o uso de enema de contraste. O tratamento padrão consiste na ressecção cirúrgica do segmento agangliónico.
Last updated: Dec 15, 2025
A doença de Hirschsprung (DH), também conhecida como aganglionose congénita ou megacólon congénito, é uma anomalia congénita do cólon causada pela falha na migração das células ganglionares derivadas da crista neural até ao cólon distal.
A fisiopatologia na DH consiste na ausência completa de células ganglionares (aganglionose), importantes na enervação intrínseca do intestino. Isto ocorre devido à falha na migração completa das células ganglionares derivadas da crista neural até aos plexos mioentéricos da parede do cólon.
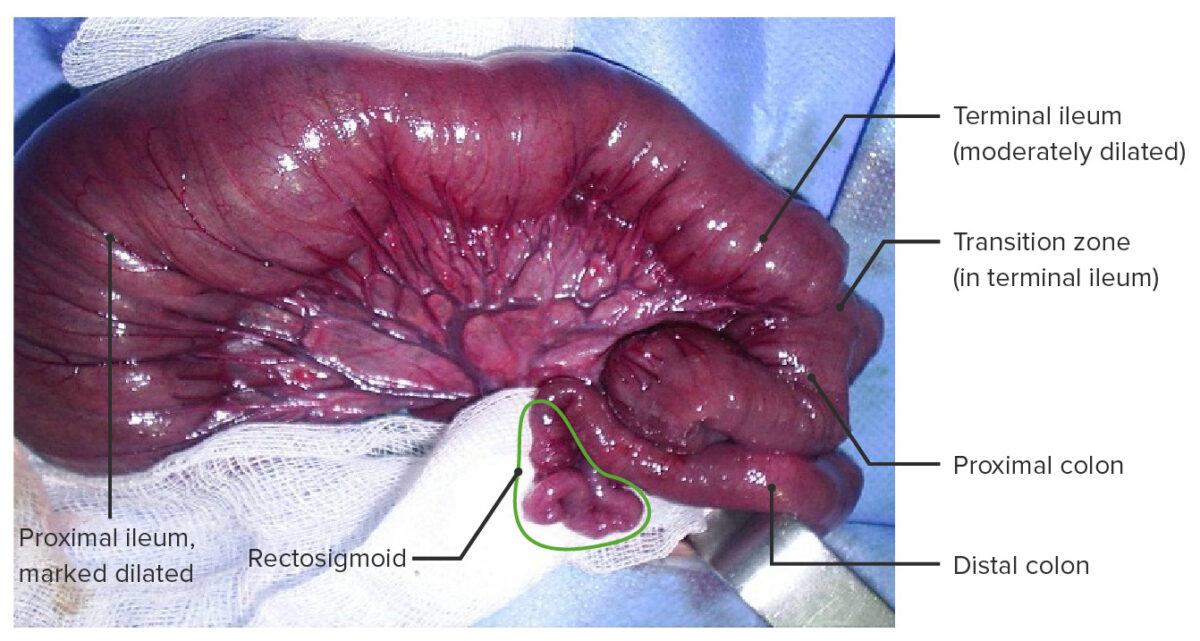
Doença de Hirschsprung com aganglionose total do cólon, incluindo num segmento pequeno do íleo terminal.
Esta forma de aganglionose ocorre em < 5% dos casos.
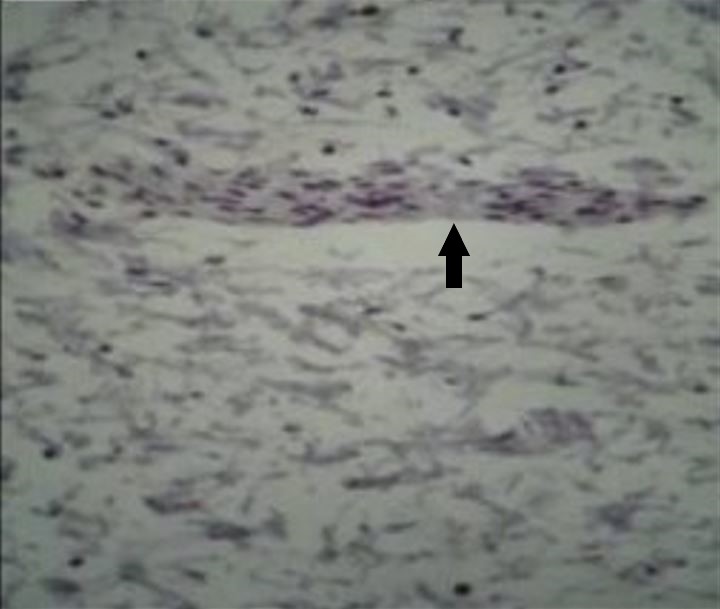
Microfotografia do segmento do intestino afetado pela doença de Hirschsprung (DH), corado com o marcador imuno-histoquímico (IHC) calretinina, onde é possível visualizar a ausência de coloração acastanhada das células ganglionares dentro de um nervo (seta) no plexo mioentérico
Imagem: “Total absence of staining after calretinin immunohistochemistry in the aganglionic segment” de Hiradfar M, Sharifi N, Khajedaluee M, Zabolinejad N, Taraz Jamshidi S. Licença: CC BY 3.0, editada por Lecturio.Na maioria dos doentes com DH o diagnóstico é feito no 1.º mês de vida. Em, aproximadamente, 10% dos casos, as crianças com doença mais MAIS Androgen Insensitivity Syndrome ligeira podem não apresentar sintomas até aos 3 anos.
Caso exista uma suspeita de DH com base nos sintomas apresentados no período neonatal ou pós-natal, são utilizados os seguintes exames:
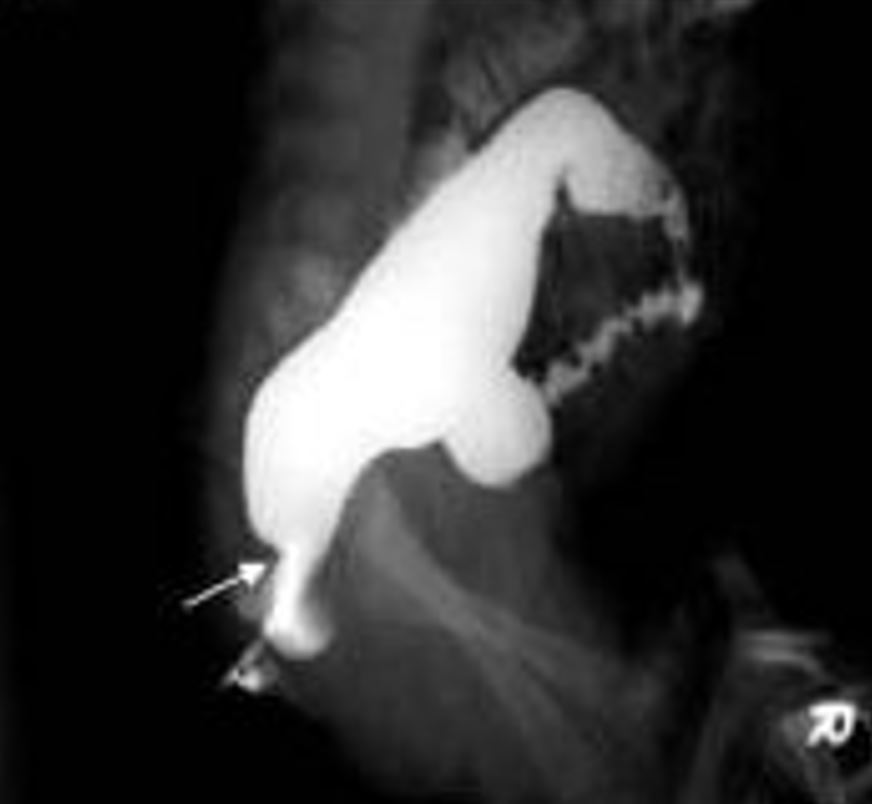
A imagem representa um estudo contrastado com enema baritado, onde é possível visualizar a doença de Hirschsprung através da seta que mostra a “zona de transição” entre o intestino normal e aganglionar
Imagem: “Contrast enema showing a CETZ at rectosigmoid, arrow” de Pratap, A., et al. Licença: CC BY 2.0, editado por Lecturio.As causas de obstrução intestinal no período neonatal podem ser diferenciadas da DH com base nas características clínicas e na presença de gânglios na biópsia retal.
Causas de obstrução intestinal em latentes mais MAIS Androgen Insensitivity Syndrome velhos e crianças:
