A deficiência de piruvato cinase (PK, pela sigla em inglês) é uma perturbação enzimática autossómica recessiva de eritrócitos que resulta numa anemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types hemolítica. A deficiência de PK é principalmente uma doença hereditária, com um defeito no geneGeneA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.Basic Terms of GeneticsPKLR, mas pode ser adquirida secundária a condições subjacentes, tais como a leucemia. As características clínicas típicas da deficiência de PK são anemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types ligeira a moderada, icterícia, atraso no crescimento, insucesso no desenvolvimento e bossa frontalFrontalThe bone that forms the frontal aspect of the skull. Its flat part forms the forehead, articulating inferiorly with the nasal bone and the cheek bone on each side of the face.Skull: Anatomy. No entanto, são possíveis apresentações maisMAISAndrogen Insensitivity Syndrome severas, especialmente em doentes maisMAISAndrogen Insensitivity Syndrome jovens. O diagnóstico envolve excluir outras causas, maisMAISAndrogen Insensitivity Syndrome comuns, de anemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types hemolítica. Os ensaios bioquímicos e/ou testesTestesGonadal Hormones genéticos podem confirmar o diagnóstico. O tratamento pode incluir tratamento de suporte, mitapivat (um ativador PK recentemente aprovado pela FDA), exsanguino-transfusão, fototerapia ou esplenectomia. O prognóstico depende da idade, estando a idade maisMAISAndrogen Insensitivity Syndrome avançada, aquando do diagnóstico, associada a um melhor prognóstico.
A deficiência de piruvato cinase (PK, pela sigla em inglês) é uma doença enzimática, autossómica recessiva, que afeta os eritrócitos, resultando na sua hemólise crónica devido a um defeito no geneGeneA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.Basic Terms of GeneticsPKLR.
Hereditárias (mutações genéticas no geneGeneA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.Basic Terms of GeneticsPKLR):
Os indivíduos homozigóticos apresentam manifestações clínicas.
Os indivíduos heterozigóticos são geralmente assintomáticos.
GeneGeneA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.Basic Terms of GeneticsPKLR localizado no cromossoma 1q21
Adquirida (secundária às condições subjacentes):
Leucemia
AnemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types sideroblástica refratária
Complicação da quimioterapia
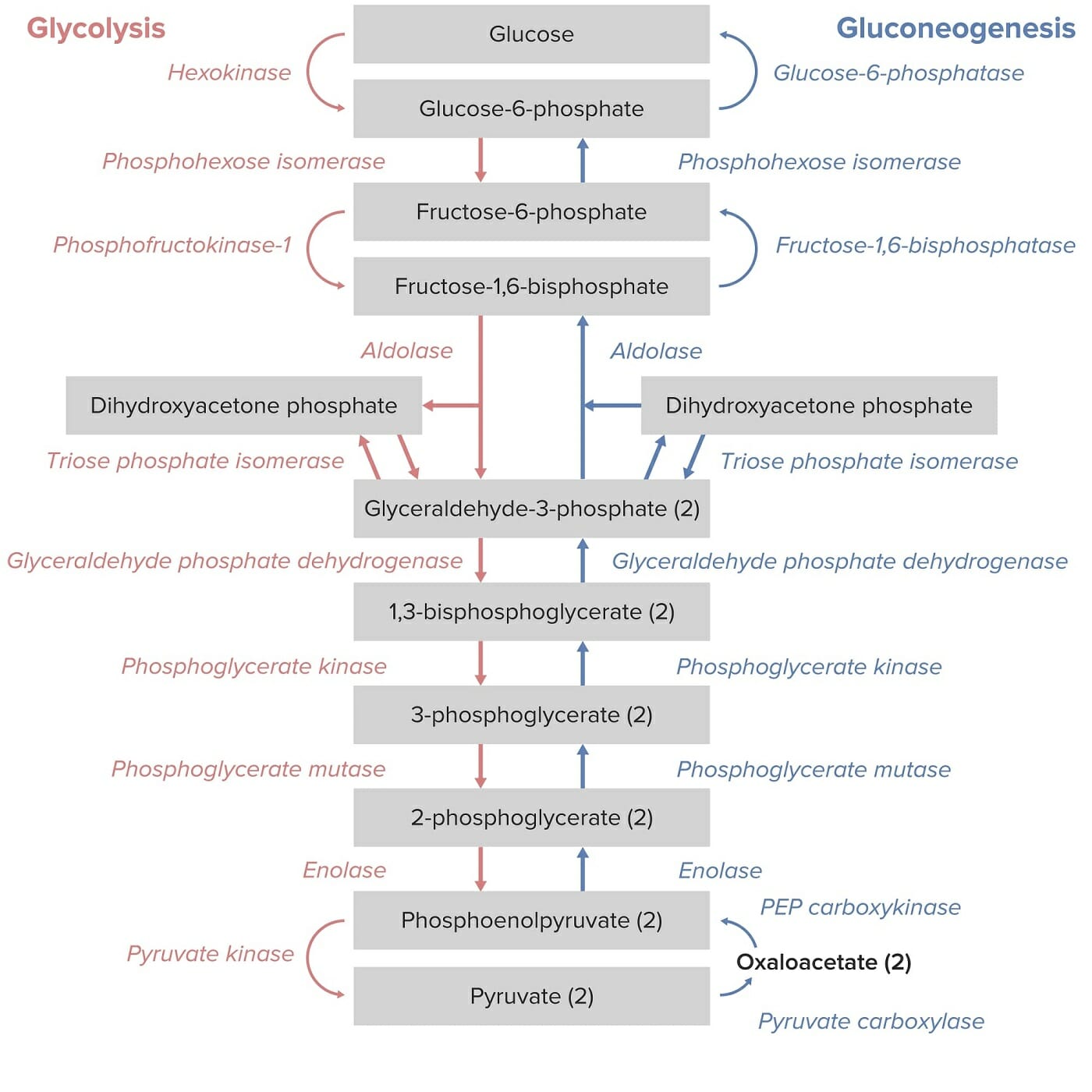
Fisiologia normal
A piruvato cinase é uma enzima necessária para a conversão de fosfoenolpiruvato (PEPPEPA monocarboxylic acid anion derived from selective deprotonation of the carboxy group of phosphoenolpyruvic acid. It is a metabolic intermediate in glycolysis; gluconeogenesis; and other pathways.Glycolysis, pela sigla em inglês) em piruvato e ATP, na via glicolítica produtora de energia.
As hemácias dependem da glicólise para a produção de ATP (não possuem mitocôndrias).
O ATP é necessário para a manutenção da função celular e integridade estrutural.
Via da glicólise e gluconeogénese: Note-se que a conversão de fosfoenolpiruvato em piruvato pela piruvato cinase é o passo final da via glicolítica.
Imagem por Lecturio.
Fisiopatologia
Efeito da deficiência de PK:
Construção de precursores dentro da via glicolítica, nomeadamente 2,3-difosfoglicerato (2,3-DPG)
↓ Produção de:
Piruvato
ATP
Lactato
Consequências da deficiência de PK:
Hemólise:
Acumulação de precursores intermediários e metabolitos glicolíticos dentro das hemácias:
↓ Plasticidade da membrana das hemácias e causa desidratação celular → Destruição prematura das hemácias no baço
A hemólise é normalmente extravascular, mas a hemólise severa pode ser intravascular.
↑ Entrega de oxigénio:
↑ 2,3-DPG → desvio para a direita na curva de dissociação do oxigénio (as hemácias têm mais dificuldade em “agarrar-se” ao O2) → resulta num ↑ da entrega de oxigénio aos tecidos periféricos, por volume de hemoglobina
Permite uma maior tolerância fisiológica à anemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types em doentes com deficiência de PK
Sobrecarga de ferro:
Eritropoiese ineficaz → ↑ produção de precursores precoces de eritrócitos
↓ Hepcidina, que leva a:
↑ Absorção de ferro
↓ Retenção de ferro em macrófagos
Apresentação Clínica e Complicações
Apresentação clínica
A idade e a gravidade da apresentação clínica dependem da extensão da hemólise e da anemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types. Além disso, a idade à apresentação pode variar.
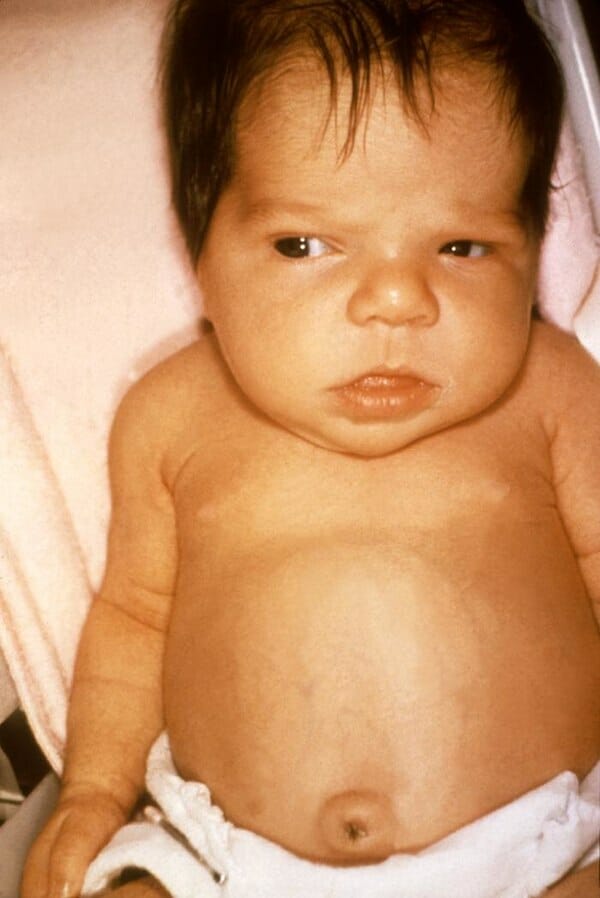
Sinais e sintomas em recém-nascidos:
Icterícia/kernicterusKernicterusA term used pathologically to describe bilirubin staining of the basal ganglia; brain stem; and cerebellum and clinically to describe a syndrome associated with hyperbilirubinemia. Clinical features include athetosis, muscle spasticity or hypotonia, impaired vertical gaze, and deafness. Nonconjugated bilirubin enters the brain and acts as a neurotoxin, often in association with conditions that impair the blood-brain barrier (e.g., sepsis). This condition occurs primarily in neonates, but may rarely occur in adults.Hyperbilirubinemia of the Newborn neonatal
Atraso do crescimento, pouco ganho de peso
Falência do desenvolvimento, letargia
Bossa frontalFrontalThe bone that forms the frontal aspect of the skull. Its flat part forms the forehead, articulating inferiorly with the nasal bone and the cheek bone on each side of the face.Skull: Anatomy
Hematopoiese extramedular cutânea
Hidrópsia fetal (devido a anemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types grave in utero)
Achados consistentes com anemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types hemolítica crónica
Palidez
Icterícia
Esplenomegalia ligeira a severa
Cálculos biliares pigmentados (bilirrubina)
Úlceras crónicas nas pernas
Uma rapariga de 6 semanas com sintomas de icterícia devido ao hipotiroidismo
Disfunção endócrina (e.g., diabetesDiabetesDiabetes mellitus (DM) is a metabolic disease characterized by hyperglycemia and dysfunction of the regulation of glucose metabolism by insulin. Type 1 DM is diagnosed mostly in children and young adults as the result of autoimmune destruction of β cells in the pancreas and the resulting lack of insulin. Type 2 DM has a significant association with obesity and is characterized by insulin resistance.Diabetes Mellitus mellitus, hipogonadismo)
Artropatias
SepsisSepsisSystemic inflammatory response syndrome with a proven or suspected infectious etiology. When sepsis is associated with organ dysfunction distant from the site of infection, it is called severe sepsis. When sepsis is accompanied by hypotension despite adequate fluid infusion, it is called septic shock.Sepsis and Septic Shock pós-esplenectomia (maior incidência em crianças)
Doença tromboembólica pós-esplenectomia (maior incidência em adultos)
Hipertensão pulmonar
Aumento do risco de anemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types aplástica perante determinadas infeções víricas (e.g., parvovírus B19)
AVC isquémico
Riscos associados a transfusões de sangue de repetição
Diagnóstico
A deficiência de piruvato cinase é rara; excluir outras causas de anemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types hemolítica é importante.
História clínica e exame objetivo
Características clínicas da anemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types hemolítica
História familiar de:
Sintomas semelhantes
Causa desconhecida de anemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types
Deficiência de piruvato cinase
Avaliação laboratorial de base
Hemograma com diferencial:
↓ Hemoglobina e hematócrito
Volume corpuscular normal ou ↑ volume corpuscular médio (VCM)
Teste direto de antiglobulina negativo (TAD; também conhecido como teste de Coombs)
Teste indireto de antiglobulina negativo
Esfregaço de sangue periférico, centrado na morfologia dos eritrócitos:
Sem achados característicos de outras causas de anemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types hemolítica hereditária.
O diagnóstico é confirmado se um doente com anemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types hemolítica/hemólise compensada apresentar anomalias nos testesTestesGonadal Hormones bioquímicos ou genéticos.
Avalia a actividade da piruvato cinase nas hemácias
Espera-se uma ↓ da atividade da piruvato cinase se deficiência de PK.
Um resultado negativo não elimina a possibilidade da deficiência de PK:
Os leucócitos e plaquetas também têm atividade PK. Se estes não forem removidos do hemolisado antes dos testesTestesGonadal Hormones, os resultados podem ser enviesados.
Se persistir a suspeita de deficiência de PK, considerar a realização de ensaios especializados ou testesTestesGonadal Hormones genéticos.
Nota: Os testesTestesGonadal Hormones bioquímicos devem ser adiados 2-3 meses após uma transfusão.
Indivíduo com hemólise ou anemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types inexplicável com um irmão com deficiência de PK
Doentes com anemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types hemolítica com um TAD negativo, quando outras causas tiverem sido descartadas
AnemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types grave dependente de transfusão, não resolvida por esplenectomia
Tratamento e Prognóstico
Tratamento
O tratamento depende da idade ao diagnóstico e da severidade.
Período intrauterino (na hidrópsia fetal devido à anemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types grave): transfusão intrauterina
Período neonatal (presença de hiperbilirrubinemia):
Fototerapia
Transfusão sanguínea
Infância até à idade adulta (presença de anemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types grave):
Transfusões de glóbulos rubros
Farmacoterapia:
Mitapivat: ativador da piruvato cinase recentemente aprovado pela FDA para o tratamento da anemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types hemolítica sintomática
Ácido fólico
Esplenectomia: indicada em casos graves, dependentes de transfusão e não responsivos ao mitapivat
Quelação do ferro (particularmente em casos que requerem tratamento de longo prazo com transfusões)
Terapêuticas em investigação:
Transplante hematopoiético de células estaminais (para doentes com anemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types grave que requerem múltiplas transfusões após esplenectomia)
Terapia genética
Monitorização
Os doentes devem ser monitorizados:
Sintomas e sinais clínicos de anemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types
Crescimento e desenvolvimento
Estudo laboratorial:
Hemograma com diferencial
Contagem de reticulócitos
Níveis de ferritina
Prognóstico
Formas suaves e moderadas de deficiência de PK: prognóstico excelente
Deficiência severa de PK: prognóstico dependente da idade
Deficiência de glucose-6-fosfato desidrogenase (G6PDG6PDPentose Phosphate Pathway): Doença hereditária recessiva ligada ao X que causa anemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types hemolítica devido à deficiência da enzima G6PDG6PDPentose Phosphate Pathway, que normalmente ajuda as hemácias a gerar NADPHNADPHNicotinamide adenine dinucleotide phosphate. A coenzyme composed of ribosylnicotinamide 5′-phosphate (nmn) coupled by pyrophosphate linkage to the 5′-phosphate adenosine 2.Pentose Phosphate Pathway, que é protetor em relação às lesões oxidativas. Apresenta sintomas de anemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types hemolítica, tais como icterícia, esplenomegalia e palidez. O diagnóstico é baseado na apresentação clínica, análise do esfregaço sanguíneo e medição do nível da enzima G6PDG6PDPentose Phosphate Pathway. O tratamento é sintomático e inclui a evicção de fatores de desencadeamento.
Doença falciforme: grupo de doenças genéticas em que uma molécula anormal de hemoglobina transforma as hemácias numa célula em forma de foice. Isto resulta numa anemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types crónica, episódios vaso-oclusivos, dor e lesão de órgão. Os doentes são suscetíveis a infeções, enfartes de vários órgãos e aplasiaAplasiaCranial Nerve Palsies da medula óssea. O envolvimento pulmonar na síndrome do tórax agudo pode ser rapidamente fatal. As células falciformes podem ser normalmente visualizadas no esfregaço periférico, mas a eletroforese da hemoglobina é necessária para o diagnóstico. O tratamento inclui tratamento sintomático, hidroxiureia, evicção de estímulos, e analgésicos.
Hiperbilirrubinemia do recém-nascido: bilirrubina sérica total elevada, que apresenta clinicamente uma descoloração amarelada da pele, escleróticas e membranas mucosas. Embora esta condição represente tipicamente um quadro de icterícia fisiológica, esta deve ser distinguida da icterícia patológica. O diagnóstico é baseado nos achados clínicos e análises sanguíneas. O tratamento pode incluir fototerapia e exsanguino-transfusão.
Esferocitose hereditária: perturbação causada por um défice da proteína citosquelética da membrana da hemácia. Isto resulta na perda de estabilidade da membrana e deformabilidade da hemácia, dando à célula uma forma esférica (esferócito). Estas células são predispostas à degradação esplénica, levando à hemólise. A condição apresenta-se com anemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types leve a moderada, hiperbilirrubinemia, reticulocitose, elevação da CHCM e esferócitos no esfregaço de sangue periférico. O tratamento inclui ácido fólico e esplenectomia (tratamento definitivo).
Referências
Bianchi, P., et al. (2018). Addressing the diagnostic gaps in pyruvate kinase deficiency: consensus recommendations on the diagnosis of pyruvate kinase deficiency. American Journal of Hematology, 94, 149‒161. https://doi.org/10.1002/ajh.25325
Grace, R. F., Barcellini, W. (2020). Management of pyruvate kinase deficiency in children and adults. Blood, 136(11), 1241–1249. https://doi.org/10.1182/blood.2019000945
Al-Samkari, H., Galactéros, F., et al. (2022). Mitapivat versus placebo for pyruvate kinase deficiency. New England Journal of Medicine, 386(15), 1432–1442. https://doi.org/10.1056/NEJMoa2116634
A Lecturio Medical complementa o teu estudo através de métodos de ensino baseados em evidência, vídeos de palestras, perguntas e muito mais – tudo combinado num só lugar e fácil de usar.
User Reviews
Details
×
Obtenha Premium para testar os seus conhecimentos
Lecturio Premium dá-lhe acesso total a todos os conteúdos e características
Obtenha Premium para ver todos os vídeos
Verifique agora o seu e-mail para obter um teste gratuito.
Crie uma conta gratuita para testar os seus conhecimentos
Lecturio Premium dá-lhe acesso total a todos os conteúdos e características - incluindo o Qbank de Lecturio com perguntas actualizadas ao estilo do board-.