Las mucopolisacaridosis, un subconjunto de las enfermedades de depósito lisosomal, son un grupo de trastornos hereditarios caracterizados por enzimas ausentes o defectuosas necesarias para descomponer las cadenas de carbohidratos llamadas glucosaminoglucanos. Estos trastornos conducen a la acumulación de glucosaminoglucanos dentro de las células, lo que da como resultado una variedad de problemas de salud. La mayoría de pacientes parecen sanos al AL Amyloidosis nacer, pero la función física y/o mental se deteriora a medida que avanza la acumulación. Con la progresión de la enfermedad, pueden verse afectados múltiples órganos. El diagnóstico se puede realizar midiendo las concentraciones de glucosaminoglucanos en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la orina y realizando ensayos enzimáticos para identificar la deficiencia enzimática. El tratamiento depende del trastorno específico, el grado de acumulación y el grado de deformidad.
Last updated: Apr 23, 2025
Las mucopolisacaridosis son un grupo de enfermedades metabólicas genéticas debidas a enzimas ausentes o defectuosas necesarias para descomponer las cadenas de carbohidratos llamadas glucosaminoglucanos.
Todas las mucopolisacaridosis se heredan y se clasifican según la deficiencia enzimática.
Las personas afectadas generalmente no se ven afectadas al AL Amyloidosis nacer, pero experimentan una progresión de la enfermedad a medida que envejecen. Las características clínicas varían según el tipo de mucopolisacaridosis.
Resulta en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum un espectro continuo de enfermedad, que se divide en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum 3 entidades clínicas según la severidad de la enfermedad (formas menos severas denominadas atenuadas):
Características clínicas distintivas (más o menos pronunciadas según la severidad):
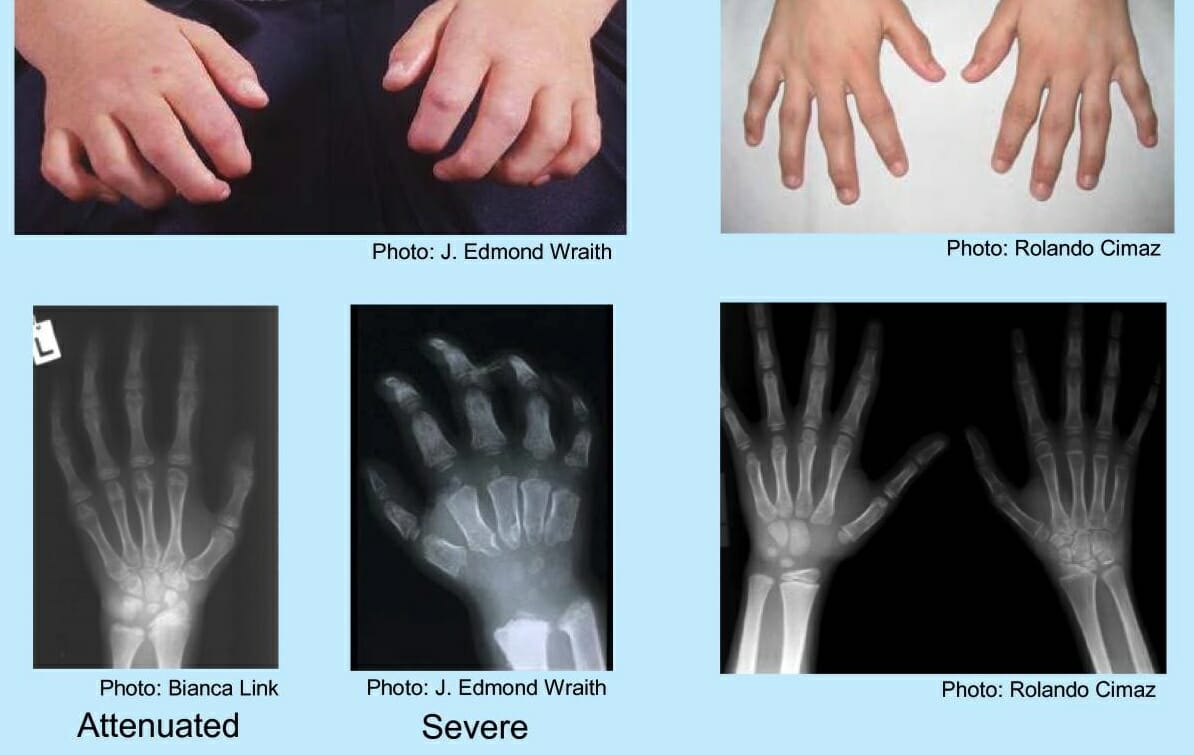
Manos de un niño con mucopolisacaridosis:
Los hallazgos incluyen una mano en “garra”, huesos metacarpianos anormales, ensanchamiento proximal de las falanges y una deformidad en forma de V en el radio y el cúbito distales.
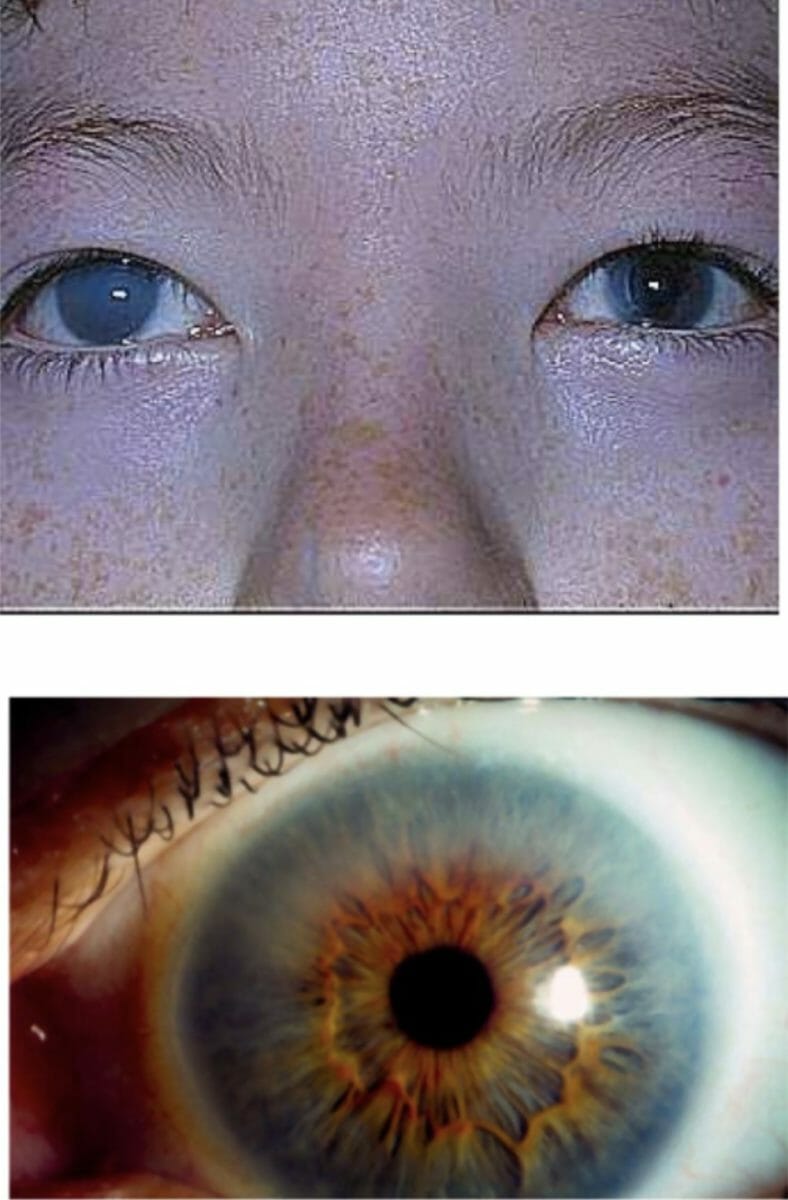
Opacidad corneal en un paciente con mucopolisacaridosis I: Este hallazgo puede ser severo, como se muestra en la imagen superior, o puede ser menos notorio, como se muestra en la imagen inferior.
Imagen: “Corneal Clouding in Patients with MPS I” por Ospedale Meyer-Reumatologia, Firenze, Italy. Licencia: CC BY 2.0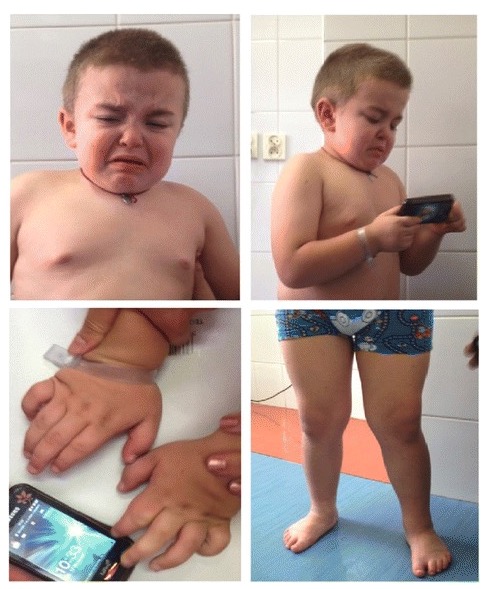
Características clínicas de la mucopolisacaridosis IIIA:
Se muestran dismorfismo facial (rasgos faciales toscos, puente nasal levemente deprimido, cejas prominentes, orejas de implantación baja, maloclusión, mejillas grandes, cabello áspero y seco, y cuello corto) y síntomas esqueléticos (genu valgo, pies en varo y manos fornidas).
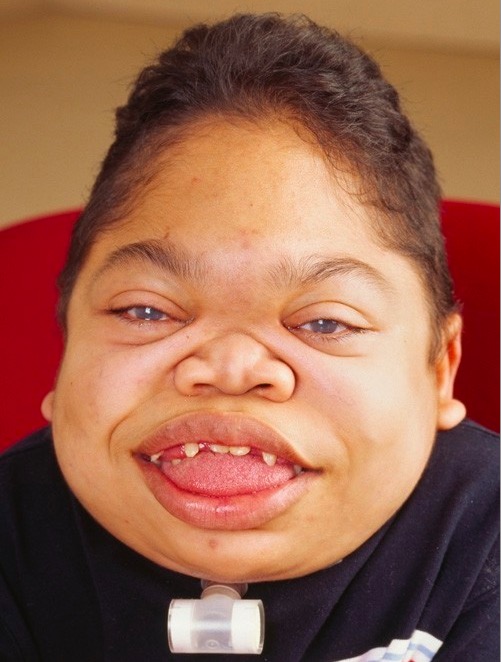
Mucopolisacaridosis VI de progresión rápida en un paciente de 16 años:
La cara muestra una facies tosca, con protuberancia frontal, lengua agrandada, labios gruesos, dentición anormal e hiperplasia gingival.
Antecedentes clínicos y examen físico:
Análisis de laboratorio:
No hay cura para la mucopolisacaridosis. La atención médica tiene como objetivo tratar las afecciones sistémicas y mejorar la calidad de vida.