La miocardiopatía dilatada (MCD) es el tipo más común de miocardiopatía no isquémica y una causa común de insuficiencia cardíaca ( IC IC Inhaled Anesthetics). La causa puede ser idiopática, familiar o secundaria a una variedad de condiciones subyacentes. La enfermedad se caracteriza por el agrandamiento de uno o ambos ventrículos y una función sistólica reducida. Los LOS Neisseria pacientes suelen presentar síntomas de IC IC Inhaled Anesthetics, como disnea, fatiga, debilidad y edema Edema Edema is a condition in which excess serous fluid accumulates in the body cavity or interstitial space of connective tissues. Edema is a symptom observed in several medical conditions. It can be categorized into 2 types, namely, peripheral (in the extremities) and internal (in an organ or body cavity). Edema periférico. Por lo general, se realizan análisis de sangre, ECG ECG An electrocardiogram (ECG) is a graphic representation of the electrical activity of the heart plotted against time. Adhesive electrodes are affixed to the skin surface allowing measurement of cardiac impulses from many angles. The ECG provides 3-dimensional information about the conduction system of the heart, the myocardium, and other cardiac structures. Electrocardiogram (ECG), radiografías, ecocardiografía, y otros estudios y procedimientos cardíacos para obtener el diagnóstico. El tratamiento incluye medicamentos utilizados para reducir la sobrecarga de volumen (e.g., diuréticos) y controlar la IC IC Inhaled Anesthetics (e.g., betabloqueadores). También pueden ser necesarios dispositivos como marcapasos y cardioversores-desfibriladores. En EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum casos severos, se requiere un trasplante cardíaco. Las complicaciones incluyen eventos tromboembólicos y muerte súbita cardíaca.
Last updated: Dec 15, 2025
La miocardiopatía dilatada es una enfermedad del músculo cardíaco:
Los LOS Neisseria síntomas pueden desarrollarse y progresar lentamente con el tiempo, o desarrollarse abruptamente. Los LOS Neisseria síntomas son similares a los LOS Neisseria observados con la IC IC Inhaled Anesthetics.
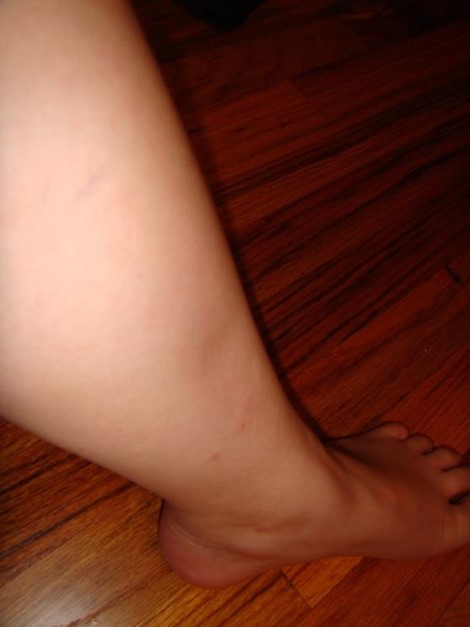
Edema con fóvea de la extremidad inferior:
La acumulación de líquido en los tejidos blandos de las extremidades inferiores puede ser un signo de IC de múltiples etiologías.
El diagnóstico se realiza principalmente a través de los LOS Neisseria antecedentes y el examen físico, exámenes de laboratorio, el ECG ECG An electrocardiogram (ECG) is a graphic representation of the electrical activity of the heart plotted against time. Adhesive electrodes are affixed to the skin surface allowing measurement of cardiac impulses from many angles. The ECG provides 3-dimensional information about the conduction system of the heart, the myocardium, and other cardiac structures. Electrocardiogram (ECG) e imagenología (confirmatorias o para excluir otras etiologías).
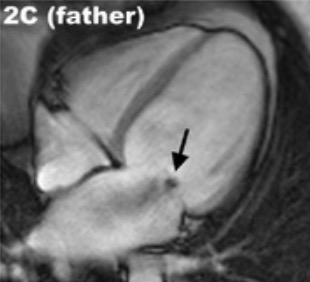
Imagenología de MCD con RM:
La miocardiopatía dilatada (MCD) con insuficiencia mitral funcional (flecha negra) se puede cuantificar y estudiar con mayor precisión gracias a las modalidades de imagen avanzadas.
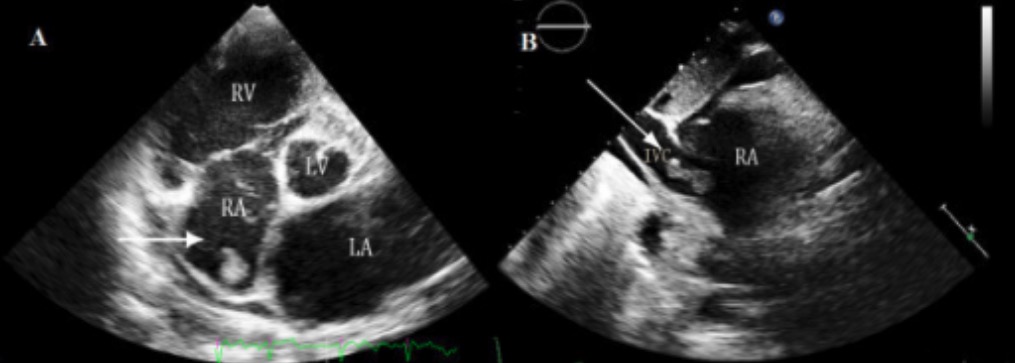
Ecocardiograma en MCD:
El ecocardiograma suele ser una herramienta de primera línea que se utiliza para evaluar la miocardiopatía y proporciona información detallada sobre el tamaño, la estructura y la función del corazón. Un ecocardiograma también puede identificar complicaciones (como el trombo en la aurícula derecha que se visualiza en esta imagen) a través de una ecocardiografía transtorácica 3D en tiempo real en un paciente con miocardiopatía dilatada.
El pronóstico es generalmente malo para las personas con esta afección.