La histiocitosis de células de Langerhans (HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have "hair-like projections" visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia) es un trastorno neoplásico clonal poco frecuente de las células dendríticas causado por mutaciones somáticas de losLOSNeisseriagenesGenesA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.DNA Types and StructureBRAF, MAP2K1, RASRASRenal artery stenosis (RAS) is the narrowing of one or both renal arteries, usually caused by atherosclerotic disease or by fibromuscular dysplasia. If the stenosis is severe enough, the stenosis causes decreased renal blood flow, which activates the renin-angiotensin-aldosterone system (RAAS) and leads to renovascular hypertension (RVH).Renal Artery Stenosis y ARAF. LosLOSNeisseria síntomas generalizados pueden incluir fiebre, fatiga y pérdida de peso. La HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have "hair-like projections" visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia pulmonar se presenta con disnea, dolorDolorInflammation torácico pleurítico y tosTOSThoracic outlet syndrome (TOS) is a broad term used for a spectrum of syndromes related to the general region of the thoracic outlet, which involves the compression or irritation of elements of the brachial plexus, subclavian artery, or subclavian vein.Thoracic Outlet Syndrome no productiva. Las manifestaciones no pulmonares de la HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have "hair-like projections" visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia dependen del órgano afectado (por ejemplo, dolorDolorInflammation óseo, endocrinopatías). El enfoque diagnóstico implica una anamnesis y una exploración física exhaustivas, con pruebas de laboratorio de referencia y pruebas de imagen (por ejemplo, radiografías). La tomografía por emisión de positrones (PETPETAn imaging technique that combines a positron-emission tomography (PET) scanner and a ct X ray scanner. This establishes a precise anatomic localization in the same session.Nuclear Imaging)-CT de cuerpo entero detecta la actividad de la enfermedad, y la biopsia de las lesiones confirma el diagnóstico. El tratamiento depende de la extensión de la enfermedad. LosLOSNeisseria métodos de tratamiento incluyen terapia localizada (por ejemplo, curetaje de la lesión para la enfermedad ósea, radioterapia) y terapia sistémica (por ejemplo, agentes quimioterapéuticos, terapia dirigida).
La histiocitosis de células de Langerhans (HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have “hair-like projections” visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia) es un trastorno neoplásico clonal poco frecuente de las células dendríticas (células de Langerhans), que intervienen enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la presentación de antígenos a las células T.
Epidemiología
Trastorno histiocítico más frecuente
Incidencia estimada:
Niños (< 15 años de edad):
5-8 casos por millón y año
Más frecuente enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum niños de 1-3 años
Adultos: 1-2 casos por millón y año
Más frecuente enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum varones que enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum mujeres
La incidencia es mayor enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la población blanca.
Fumar enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum exceso aumenta el riesgo de HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have “hair-like projections” visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia pulmonar monosistémica.
Clasificación
Tanto la Histiocyte Society como la OMS disponen de clasificaciones para losLOSNeisseria trastornos histiocíticos.
La clasificación de la Histiocyte Society incluye trastornos malignos y no malignos.
La clasificación de la OMS solo aborda losLOSNeisseria trastornos malignos.
Clasificación de la Histiocyte Society
La Histiocyte Society divide losLOSNeisseria trastornos histiocíticos enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum 5 categorías basadas enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum lo siguiente:
Características clínicas
Características histológicas
Características inmunofenotípicas
Características moleculares
Tabla: Clasificación de la Histiocyte Society
Grupo de trastornos histiocíticos
Trastornos
Grupo de Langerhans (L)
Histiocitosis de células de Langerhans (HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have “hair-like projections” visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia)
Enfermedad de Erdheim-Chester
Mixto LCH/Erdheim-Chester
Histiocitosis de células indeterminadas
Xantogranuloma juvenil extracutáneo
Grupo cutáneo y mucocutáneo (C)
Xantogranuloma juvenil
Xantogranuloma del adulto
Enfermedad cutánea de Rosai-Dorfman
Otros trastornos histiocíticos localizados enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la piel o las mucosas que no cumplen losLOSNeisseria criterios diagnósticos de la HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have “hair-like projections” visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia
Grupo de la enfermedad de Rosai-Dorfman (R)
Enfermedad de Rosai-Dorfman
Histiocitosis no cutáneas diversas que no cumplen losLOSNeisseria criterios diagnósticos de la HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have “hair-like projections” visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia
Grupo de histiocitosis malignas (M)
Histiocitosis malignas primarias con afectación multisistémica
Histiocitosis malignas secundarias a otros linfomas y leucemias
Grupo de linfohistiocitosis hemofagocítica (H)
Linfohistiocitosis hemofagocítica primaria
Síndromes de activación macrofágica (SAMSAMAnterior displacement of the mitral valve during systole.Hypertrophic Cardiomyopathy)
Etiología y Fisiopatología
Etiología
La HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have “hair-like projections” visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia está causada por mutaciones somáticas de genesGenesA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.DNA Types and Structure que regulan la vía de señalización MAPK/ERK, como:
BRAF
MAP2K1
RASRASRenal artery stenosis (RAS) is the narrowing of one or both renal arteries, usually caused by atherosclerotic disease or by fibromuscular dysplasia. If the stenosis is severe enough, the stenosis causes decreased renal blood flow, which activates the renin-angiotensin-aldosterone system (RAAS) and leads to renovascular hypertension (RVH).Renal Artery Stenosis
ARAF
Fisiopatología
Mutaciones genéticas → activación de la vía MAPK → expansión clonal de precursores mieloides.
Las células dendríticas proliferan y se acumulan debido a la proliferación clonal impulsada por la vía MAPK.
A continuación, las células dendríticas que proliferan anormalmente se infiltran enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum ≥ 1 órgano → manifestaciones clínicas
La HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have “hair-like projections” visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia puede presentarse como un trastorno monosistémico o multisistémico.
LCH monosistémica:
LosLOSNeisseria síntomas sistémicos suelen estar ausentes.
HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have “hair-like projections” visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia multisistémica:
Afecta a ≥ 2 órganos
La combinación de órganos implicados varía, pero puede incluir:
Hueso
Piel
Hipotálamo-hipofisario
Ganglios linfáticos
Pulmones
SNC
Hígado
Bazo
Mucosa oral
EnENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum niños menores de 3 años, la HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have “hair-like projections” visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia suele ser una enfermedad multisistémica.
HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have “hair-like projections” visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia pulmonar
Enfermedad pulmonar histiocítica más frecuente
Anteriormente conocida como granuloma eosinofílico de pulmón
Más común enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum adultos
Asociada alALAmyloidosis tabaquismo (el 90% de losLOSNeisseria pacientes son ex fumadores o fumadores actuales)
Se evidencian síntomas constitucionales (por ejemplo, fiebre, pérdida de peso) enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el 20% de losLOSNeisseria casos.
HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have “hair-like projections” visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia no pulmonar
LosLOSNeisseria signos y síntomas dependen del número y la localización de las zonas afectadas.
Tabla: Presentación clínica de la HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have “hair-like projections” visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia por localización
Sistema
Manifestaciones (pueden incluir cualquiera de las siguientes):
TosTOSThoracic outlet syndrome (TOS) is a broad term used for a spectrum of syndromes related to the general region of the thoracic outlet, which involves the compression or irritation of elements of the brachial plexus, subclavian artery, or subclavian vein.Thoracic Outlet Syndrome no productiva
Secreción del oído, pérdida de audición (hueso temporal)
Cutáneo
Puede producirse una amplia gama de erupciones:
Erupción eccematosa
Pápulas marrón-púrpura
Lesiones ulcerosas
También pueden ser pustulosas, purpúricas, petequiales o vesiculares
Pueden tener descamación, costras o supuración
Decoloración y endurecimiento de las uñas
Puede verse afectada cualquier parte del cuerpo.
Oral
Hipertrofia gingival
Enfermedad periodontal
Masa intraoral
Úlceras mucosas
Dientes flojos
Hematológico
AnemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types
Sangre enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum las heces
Hígado/bazo
Distensión abdominal
Ictericia
Prurito
Hepatomegalia y/o esplenomegalia
Endocrino
Polidipsia
Poliuria
Retraso del crecimiento y de la pubertad
Aumento de peso (afectación hipofisaria)
Síntomas de hipotiroidismo (afectación de la glándula tiroides)
Sistema nervioso central
Movimientos corporales descoordinados
AtaxiaAtaxiaImpairment of the ability to perform smoothly coordinated voluntary movements. This condition may affect the limbs, trunk, eyes, pharynx, larynx, and other structures. Ataxia may result from impaired sensory or motor function. Sensory ataxia may result from posterior column injury or peripheral nerve diseases. Motor ataxia may be associated with cerebellar diseases; cerebral cortex diseases; thalamic diseases; basal ganglia diseases; injury to the red nucleus; and other conditions.Ataxia-telangiectasia
Deficiencias hormonales hipofisarias (por ejemplo, diabetesDiabetesDiabetes mellitus (DM) is a metabolic disease characterized by hyperglycemia and dysfunction of the regulation of glucose metabolism by insulin. Type 1 DM is diagnosed mostly in children and young adults as the result of autoimmune destruction of β cells in the pancreas and the resulting lack of insulin. Type 2 DM has a significant association with obesity and is characterized by insulin resistance.Diabetes Mellitus insípida central)
Auricular
Secreción
Erupción roja y pruriginosa enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el conducto auditivo externo
TosTOSThoracic outlet syndrome (TOS) is a broad term used for a spectrum of syndromes related to the general region of the thoracic outlet, which involves the compression or irritation of elements of the brachial plexus, subclavian artery, or subclavian vein.Thoracic Outlet Syndrome
Disnea
Cianosis
Síndrome de la vena cava superior
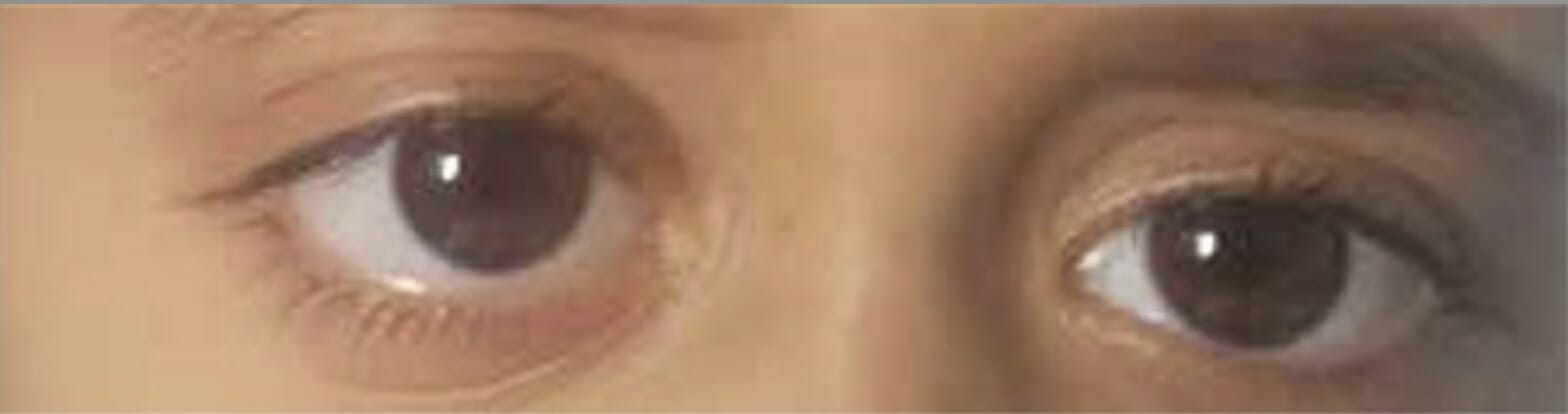
Exoftalmos derecho por afectación de la pared orbitaria por histiocitosis de células de Langerhans
Imagen: “Proptosis due to Langerhans Cell” por Turkish Journal of Ophthalmology. Licencia: CC BY 2.5
Masa mal definida (flecha) en la pared orbitaria lateral debida a una histiocitosis de células de Langerhans.
Imagen: “LCH presented with ill-defined mass in lateral orbital wall” por Departamento de Oftalmología, Sri Sankaradeva Nethralaya, Guwahati. Licencia: CC BY 2.0
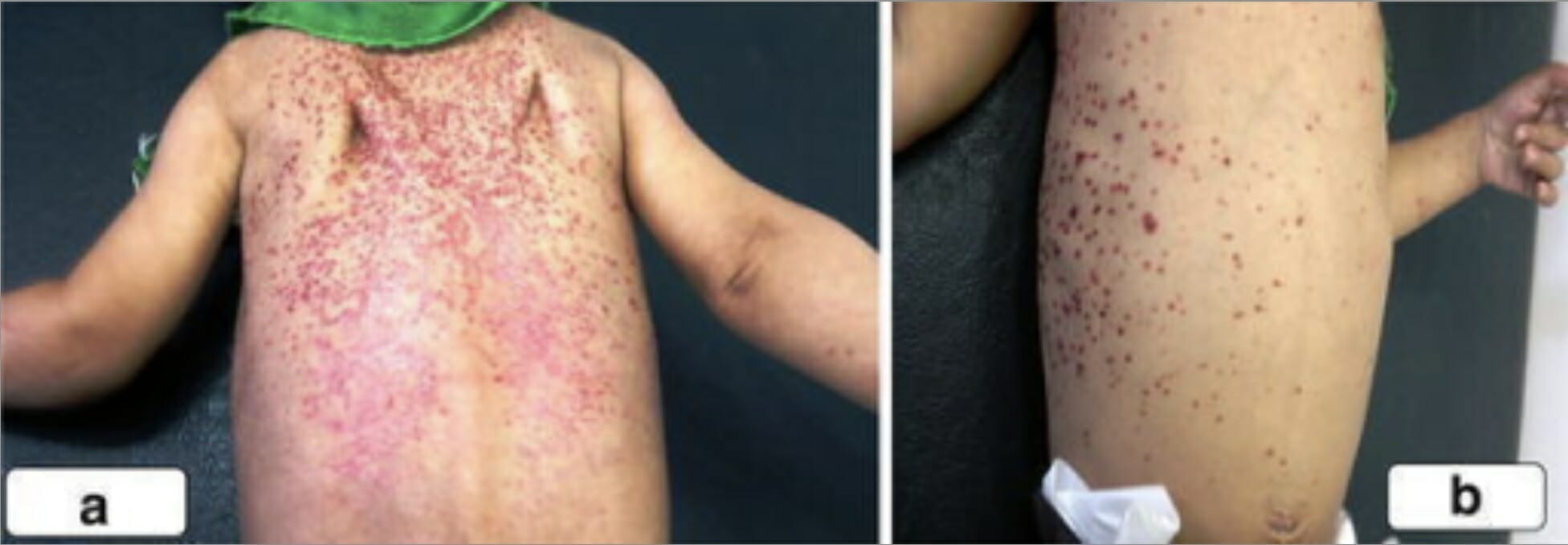
Erupción eritematosa descamativa en la espalda, los hombros y el pecho de un niño con histiocitosis de células de Langerhans.
Imagen: “A scaly, erythematous rash on the back of the second boy spread to his shoulders (a) and upper chest wall (b)” por la Unidad de Cirugía Pediátrica, Al Diwaniya General Teaching Hospital, Al Qadisiya, Iraq. Licencia: CC BY 4.0
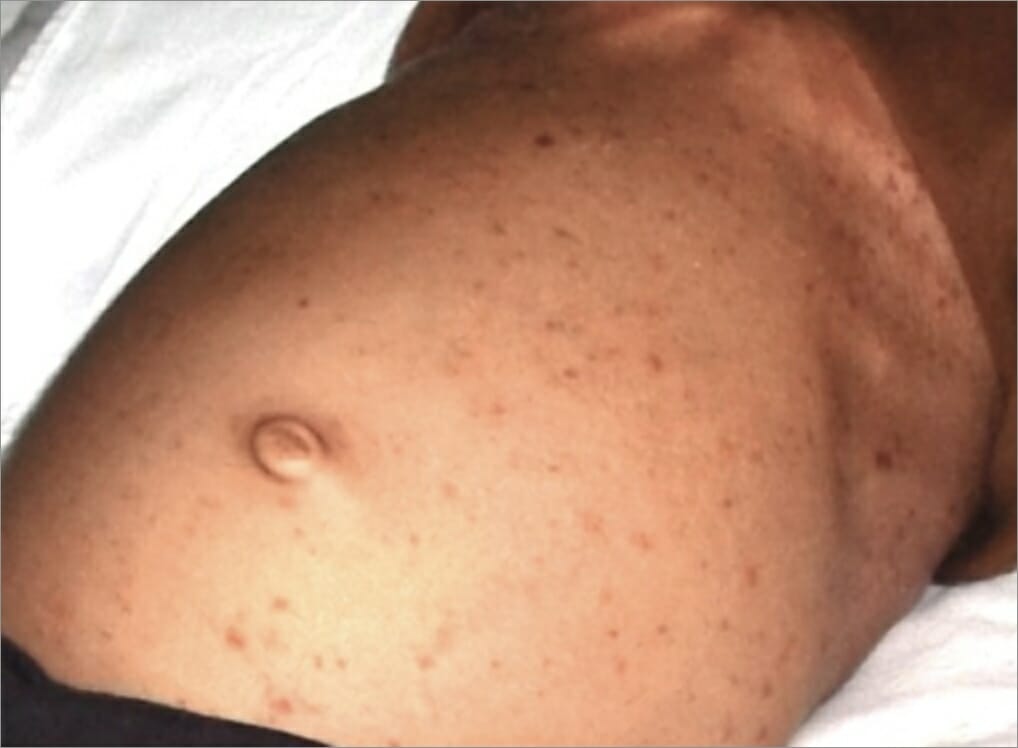
Erupción de pápulas en el estómago de un individuo como manifestación de histiocitosis de células de Langerhans.
Imagen: “After hospitalization” por el Departamento de Medicina Oral y Radiología, Jaipur Dental College, Jaipur, Rajasthan, India. Licencia: CC BY 3.0
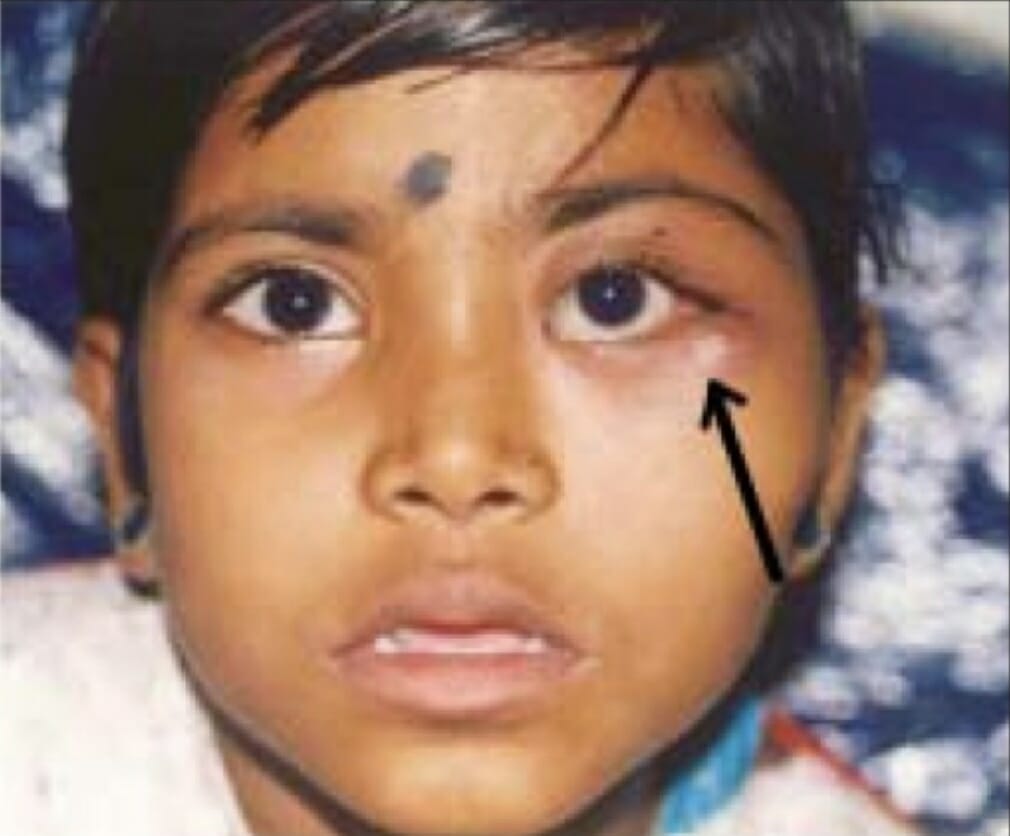
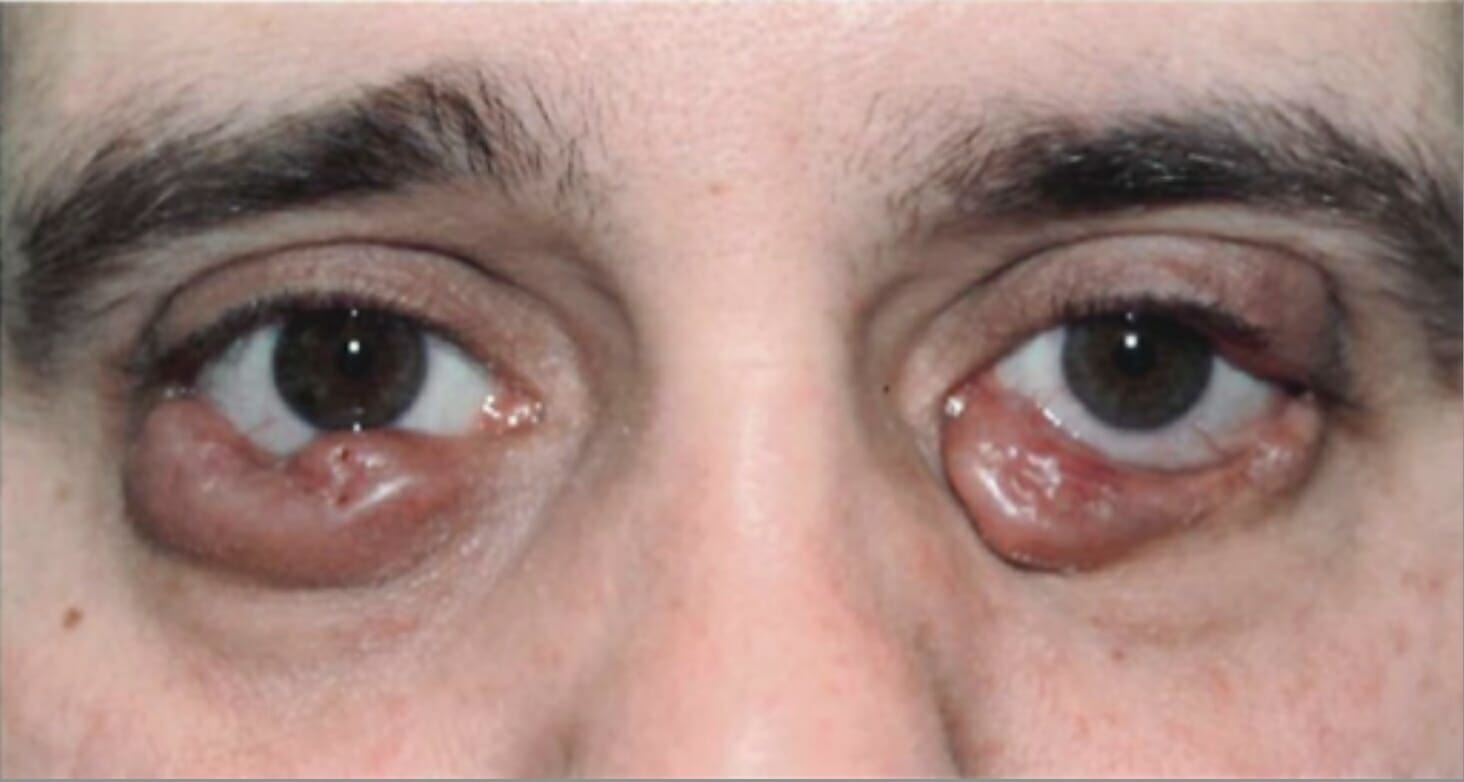
Tumores bilaterales del párpado inferior debidos a histiocitosis de células de Langerhans
Imagen: “Before radiation treatment“, por King’s College de Londres. Licencia: CC BY 3.0
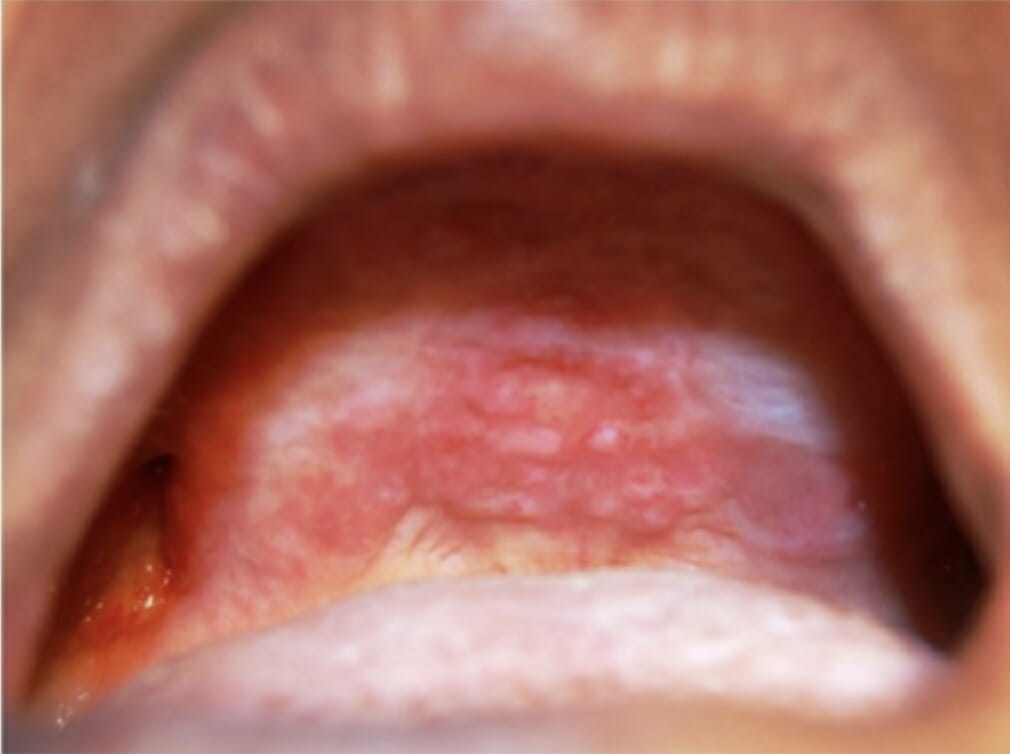
Lesiones orales en el paladar duro y la cresta alveolar maxilar como manifestación de histiocitosis de células de Langerhans
Imagen: “Oral lesions in the hard palate and maxillary alveolar ridge” por el Departamento de Medicina Oral y Radiología, M S Ramaiah Dental College and Hospital, Bangaluru. Licencia: CC BY 2.5
Complicaciones
Las complicaciones dependen del sistema orgánico afectado, pero pueden incluir:
Neumotórax
Enfermedad vascular pulmonar
Disfunción pulmonar crónica
Fractura ósea patológica
Cirrosis hepática
Perforación intestinal
Hemorragia digestiva
Neoplasias malignas secundarias como la leucemia linfoblástica aguda
Ceguera (poco frecuente)
Diagnóstico
Estudios de laboratorio
LosLOSNeisseria estudios de laboratorio suelen mostrar hallazgos coherentes con losLOSNeisseria sistemas orgánicos implicados y ayudan a descartar otros diagnósticos (la lista no es exhaustiva):
Exámenes generales:
Hemograma completo (con diferencial) → evaluar la afectación de la médula ósea:
AnemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types
Trombocitopenia
Leucopenia
Tiempo de protrombina (TP) y tiempo de tromboplastina parcial activado (TTPa) → coagulopatía (especialmente con afectación hepática).
Electrolitos → pueden demostrar anomalías relacionadas con trastornos endocrinos.
Pruebas de función renal → evaluar la afectación renal.
Perfil hepático → ↑ con afectación hepatobiliar.
Una evaluación endocrina detallada puede incluir:
Hormona estimulante de la tiroides (TSH), T4T4The major hormone derived from the thyroid gland. Thyroxine is synthesized via the iodination of tyrosines (monoiodotyrosine) and the coupling of iodotyrosines (diiodotyrosine) in the thyroglobulin. Thyroxine is released from thyroglobulin by proteolysis and secreted into the blood. Thyroxine is peripherally deiodinated to form triiodothyronine which exerts a broad spectrum of stimulatory effects on cell metabolism.Thyroid Hormones libre
Prolactina y factor de crecimiento similar a la insulina 1 (IGF-1)
Hormona foliculoestimulante (FSHFSHA major gonadotropin secreted by the adenohypophysis. Follicle-stimulating hormone stimulates gametogenesis and the supporting cells such as the ovarian granulosa cells, the testicular sertoli cells, and leydig cells. Fsh consists of two noncovalently linked subunits, alpha and beta. Within a species, the alpha subunit is common in the three pituitary glycoprotein hormones (TSH, LH, and FSH), but the beta subunit is unique and confers its biological specificity.Menstrual Cycle) y hormona luteinizante (LHLHA major gonadotropin secreted by the adenohypophysis. Luteinizing hormone regulates steroid production by the interstitial cells of the testis and the ovary. The preovulatory luteinizing hormone surge in females induces ovulation, and subsequent luteinization of the follicle. Luteinizing hormone consists of two noncovalently linked subunits, alpha and beta. Within a species, the alpha subunit is common in the three pituitary glycoprotein hormones (TSH, LH, and FSH), but the beta subunit is unique and confers its biological specificity.Menstrual Cycle)
Testosterona (hombres)
EstradiolEstradiolThe 17-beta-isomer of estradiol, an aromatized C18 steroid with hydroxyl group at 3-beta- and 17-beta-position. Estradiol-17-beta is the most potent form of mammalian estrogenic steroids.Noncontraceptive Estrogen and Progestins (mujeres)
Imagenología y estudios auxiliares adicionales
Radiografía basal enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la presentación inicial:
Radiografía de tórax → imagen inicial para síntomas pulmonares:
Nódulos mal definidos o estrellados
Patrón reticulonodular bilateral difuso (zonas media y superior)
Quistes o patrón enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum panal enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la zona superior
Volumen pulmonar conservado
Conservación del ángulo costofrénico
Radiografía del hueso/articulación afectado: Las lesiones suelen ser osteolíticas (aspecto “perforado”).
PET-TC de cuerpo entero con fluorodesoxiglucosa (FDG):
La HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have “hair-like projections” visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia es ávida de FDG, por lo que la PET-TC detecta la afectación orgánica.
Superior a la radiografía, la TC o la RM enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la detección de lesiones óseas de HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have “hair-like projections” visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia
Realizada enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum pacientes ≥ 2 años de edad.
Recomendada para el estadiaje
Otros estudios dependen de losLOSNeisseria lugares afectados:
RM cerebral con contraste de gadolinio:
Para afectación del SNC
Detecta lesiones de la masa craneal
Útil para evaluar la hipófisis
TC:
Puede evaluar la afectación gastrointestinal, hepática, esplénica, mediastínica y pulmonar.
EnENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have “hair-like projections” visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia pulmonar se observan opacidades reticulonodulares y quistes (zonas media y superior).
Pruebas de función pulmonar:
Evaluan la afectación pulmonar
Pueden ser normales enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum enfermedad temprana
Pueden mostrar una capacidad de difusión reducida con anomalías obstructivas, restrictivas o mixtas.
Ultrasonido: puede utilizarse enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la evaluación de hígado/tiroides.
Hallazgos de PET en histiocitosis de células de Langerhans pulmonar nodular: Las imágenes de TC torácica de los paneles superior e inferior izquierdos muestran múltiples nódulos pulmonares. Las imágenes PET correspondientes en los paneles superior e inferior de la derecha muestran la captación PET. Los nódulos pulmonares de mayor tamaño (puntas de flecha en las imágenes de TC) muestran una intensa captación de PET, mientras que otros nódulos (flechas más pequeñas en las imágenes de TC) son negativos para PET.
Imagen: “PET findings in nodular PLCH”, por la División de Medicina Pulmonar y Cuidados Críticos, Mayo Clinic, Rochester, MN, EE.UU. Licencia: CC BY 2.0
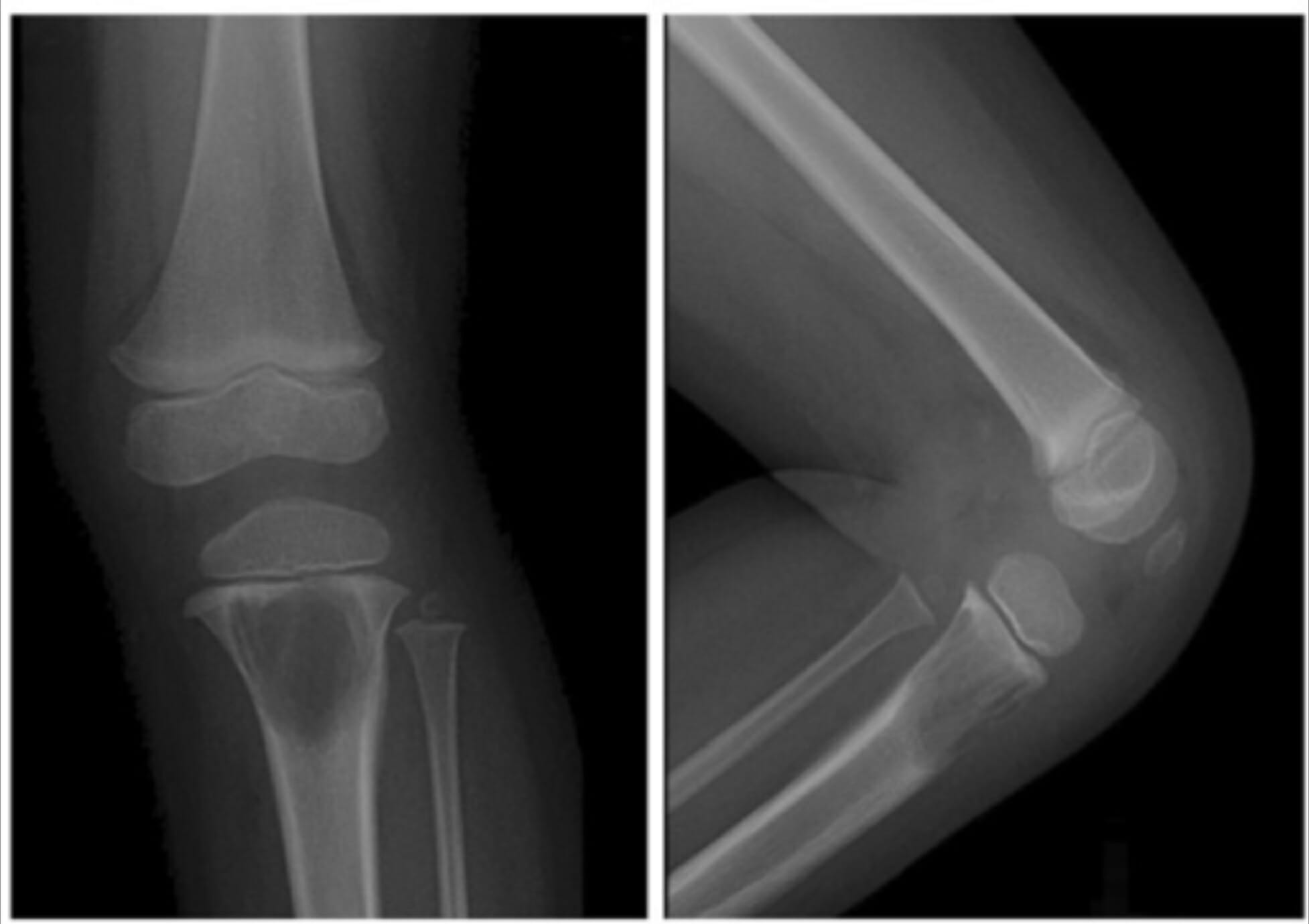
Radiografías de la rodilla izquierda de una niña que muestran una gran lesión lítica en la metáfisis proximal de la tibia debida a una histiocitosis de células de Langerhans.
Imagen: “A 2-year-old girl with left knee pain and a medullary lytic lesion in the proximal tibial metaphysis” por el Departamento de Radiología, Tri-Service General Hospital, National Defense Medical Center, Taipei, Taiwan. Licencia: CC BY 4.0
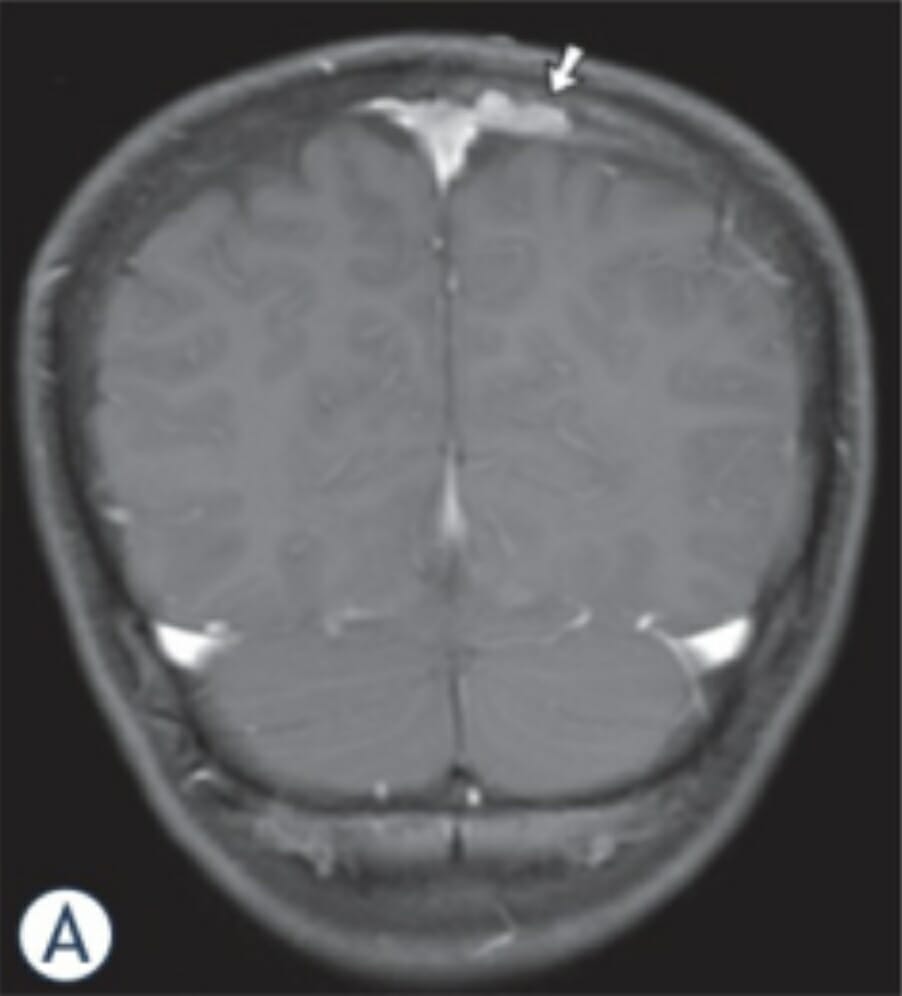
Imágenes de RM en un niño de 11 años con HCL: La imagen coronal de RM potenciada en T1 revela una masa ósea (flecha) combinada con afectación epidural y subdural a lo largo del lado izquierdo del seno sagital superior.
Imagen: “MR-images in an 11-year-old boy with LCH” por el Departamento de Neurorradiología, Universidad Johann Wolfgang Goethe, Frankfurt/Main, Alemania. Licencia: CC BY 3.0
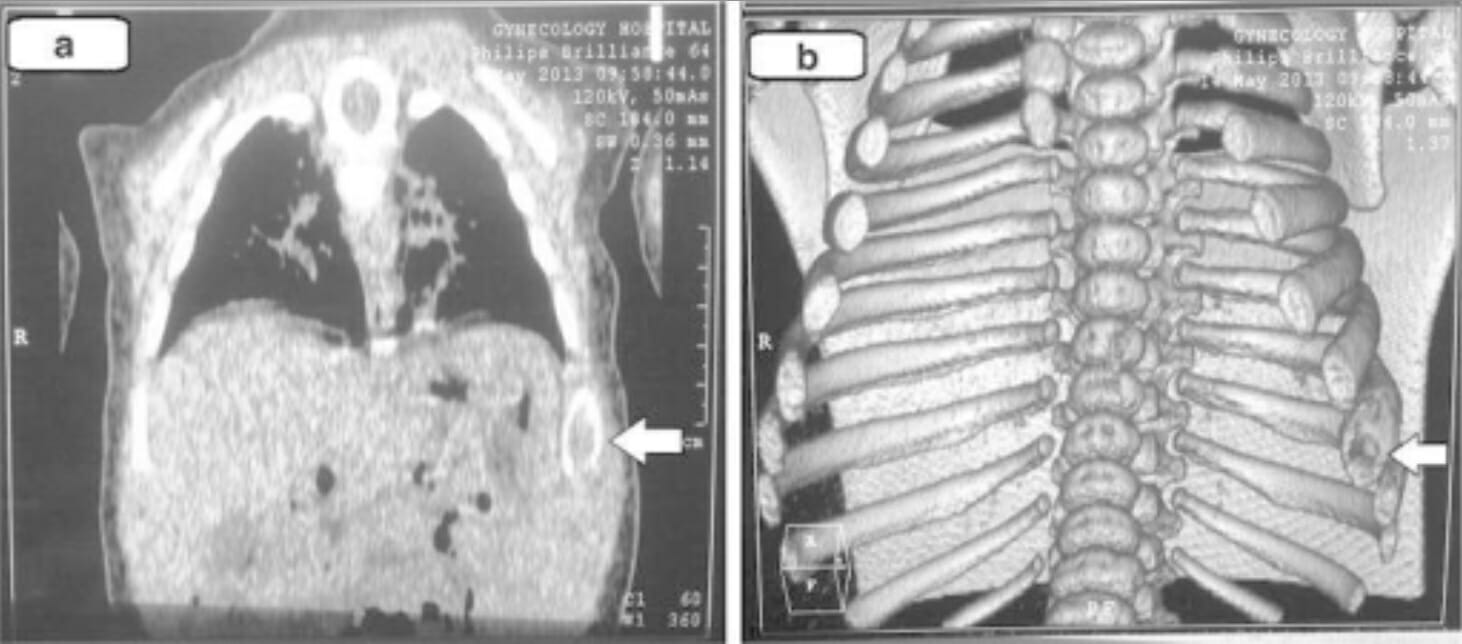
Hallazgos de la TC torácica (e imágenes tridimensionales) en la histiocitosis de células de Langerhans: Estas imágenes muestran cambios líticos en la 8va costilla izquierda (flechas).
Imagen: “Chest computed tomography” por la Unidad de Cirugía Pediátrica, Hospital General Docente Al Diwaniya, Al Qadisiya, Irak. Licencia: CC BY 4.0
Patología
Examen de la médula ósea (especialmente con un hemograma anormal)
Biopsia de la lesión/tumorTumorInflammation recomendada enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum todos losLOSNeisseria casos:
Ejemplos:
Lesión ósea osteolítica
Lesión cutánea
Lavado broncoalveolar
Biopsia de pulmón
Las características histopatológicas varían enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum función del lugar de la biopsia, mostrando tejido:
Células de Langerhans (núcleos plegados o estriados, con citoplasma moderadamente abundante)
Diversos grados de fibrosisFibrosisAny pathological condition where fibrous connective tissue invades any organ, usually as a consequence of inflammation or other injury.Bronchiolitis Obliterans
Atipia citológica enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum células neoplásicas
La inmunohistoquímica → puede ser positiva para:
CD1a
CD207 (langerina)
S100S100A family of highly acidic calcium-binding proteins found in large concentration in the brain and believed to be glial in origin. They are also found in other organs in the body. They have in common the ef-hand motif (ef hand motifs) found on a number of calcium binding proteins. The name of this family derives from the property of being soluble in a 100% saturated ammonium sulfate solution.Acoustic Neuroma
Genética:
Prueba de mutación BRAF V600E (positiva enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum > 50%)
Considerar la secuenciación de nueva generación para la vía de señalización MAPK-ERK
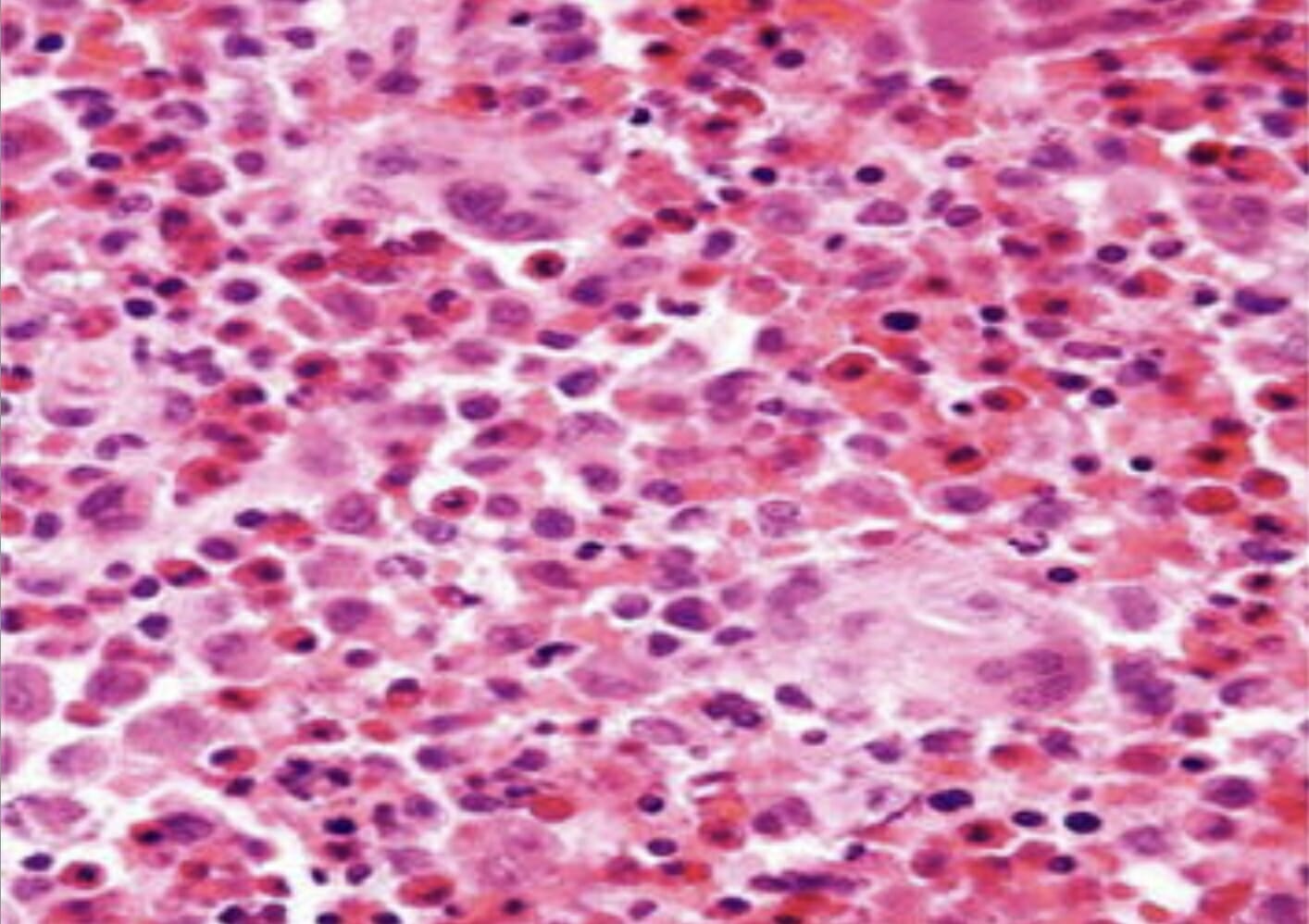
Histopatología de biopsia cutánea (hematoxilina y eosina × 100) que muestra agregados de células histiocíticas con abundante citoplasma eosinófilo y granular.
Imagen: “Skin biopsy histopathology” por la Unidad de Cirugía Pediátrica, Al Diwaniya General Teaching Hospital, Al Qadisiya, Iraq. Licencia: CC BY 4.0
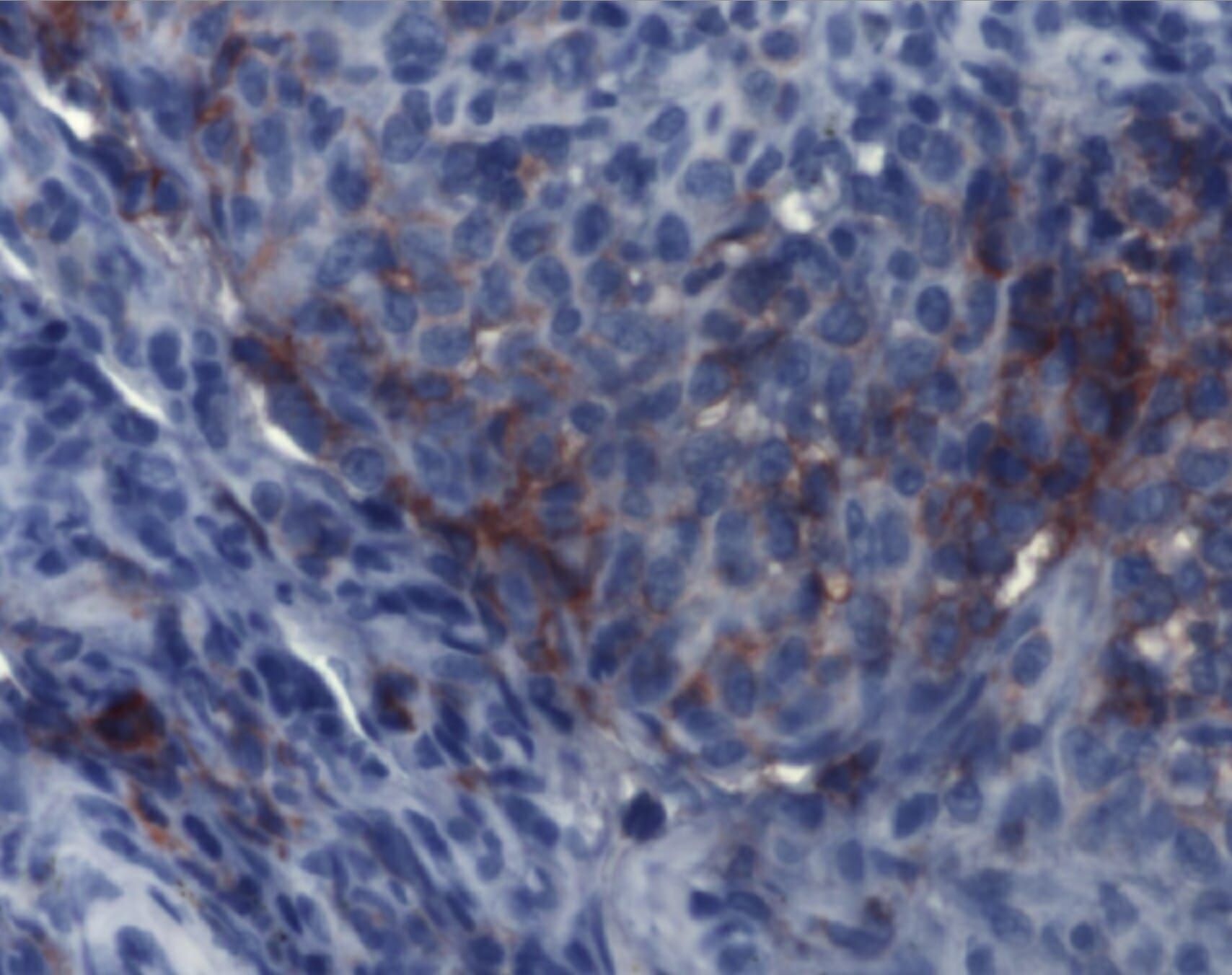
La inmunohistoquímica demuestra hallazgos compatibles con histiocitosis de células de Langerhans. Los agregados de células de Langerhans son detectables mediante anticuerpos monoclonales contra CD1a.
Imagen: “Immunohistochemistry of Langerhans cell histiocytosis” por el Instituto de Patología y Neuropatología, University Hospital Essen, University of Duisburg-Essen, Germany. Licencia: CC BY 2.0
Tratamiento y Pronóstico
La elección del tratamiento de la HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have “hair-like projections” visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia se basa enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el tipo de HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have “hair-like projections” visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia, la presentación clínica, losLOSNeisseria órganos afectados, la extensión de la afectación y la función orgánica. El tratamiento suele estar dirigido por un oncólogo (y potencialmente por otros especialistas).
Tratamiento de la HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have “hair-like projections” visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia pulmonar
Tratamiento de soporte:
Cese de tabaquismo
Evaluación periódica de la función pulmonar
Broncodilatadores inhalados y/o glucocorticoides para la limitación reversible del flujo aéreo
Rehabilitación pulmonar enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum pacientes con disnea de esfuerzo
Vacunación antineumocócica y de la influenzaInfluenzaInfluenza viruses are members of the Orthomyxoviridae family and the causative organisms of influenza, a highly contagious febrile respiratory disease. There are 3 primary influenza viruses (A, B, and C) and various subtypes, which are classified based on their virulent surface antigens, hemagglutinin (HA) and neuraminidase (NA). Influenza typically presents with a fever, myalgia, headache, and symptoms of an upper respiratory infection. Influenza Viruses/Influenza estacional
HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have “hair-like projections” visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia pulmonar sintomática/progresiva:
Glucocorticoides sistémicos
Opciones de quimioterapia:
Cladribina
Citarabina
Tratamiento de la HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have “hair-like projections” visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia no pulmonar
Si está asintomático o sin disfunción orgánica, puede considerarse la observación.
Afectación multisistémica enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum niños:
Vinblastina
Prednisona
Afectación multisistémica enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum adultos:
Opciones de quimioterapia:
citarabina
cladribina
Terapia dirigida para mutaciones genéticas conocidas:
No se utiliza enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum niños
El tratamiento con bifosfonatos puede reducir el dolorDolorInflammation.
Lesiones óseas múltiples:
Terapia sistémica (quimioterapia y terapias dirigidas similares a las utilizadas para la afectación multisistémica)
Tratamientos complementarios:
Cirugía
Radioterapia
Dermatológicos:
La HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have “hair-like projections” visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia solo cutánea puede remitir espontáneamente o evolucionar a HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have “hair-like projections” visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia multisistémica.
Pacientes asintomáticos: observación
Sintomática o con afectación cutánea extensa:
Esteroides tópicos o mostaza nitrogenada
Metotrexato, que puede combinarse con otros agentes inmunosupresores (por ejemplo, prednisolona, 6-mercaptopurina, hidroxiurea).
Lenalidomida
Talidomida
Pronóstico
La evolución clínica y el pronóstico de losLOSNeisseria pacientes con HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have “hair-like projections” visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia son variables:
Afectación de un único ganglio o enfermedad cutánea aislada
Ausencia de lesiones 1 año después del seguimiento
HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have “hair-like projections” visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia pulmonar (sin tabaquismo)
HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have “hair-like projections” visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia con mutaciones BRAF (mayor probabilidad de recaída)
Afectación de órganos de alto riesgo (por ejemplo, SNC, médula ósea, hígado, bazo)
La HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have “hair-like projections” visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia focal crónica puede evolucionar a enfermedad multifocalMultifocalRetinoblastoma o diseminada.
Diagnóstico Diferencial
Mieloma múltiple: neoplasia enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la que las células plasmáticas proliferan de forma anormal enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la médula ósea, provocando el desplazamiento de las líneas celulares hematopoyéticas. La presentación clínica puede incluir dolorDolorInflammation óseo, fracturas patológicas y signos de insuficiencia renal. Las lesiones óseas osteolíticas pueden verse enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum las imágenes. Las características diagnósticas distintivas son la proteína monoclonal enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum suero y orina, la histología (por ejemplo, células plasmáticas con abundante citoplasma basófilo) y el inmunofenotipo. El tratamiento incluye corticosteroides, quimioterapia y/o agentes inmunomoduladores.
Enfermedad de Erdheim-Chester: a menudo una enfermedad del adulto, esta enfermedad es un trastorno histiocítico multisistémico. LosLOSNeisseria infiltrados xantogranulomatosos afectan a ≥ 1 sistema orgánico (por ejemplo, piel, pulmón, hueso, cerebro, hipófisis, retroperitoneo, sistema cardiovascular). Se observa osteosclerosisOsteosclerosisAn abnormal hardening or increased density of bone tissue.Paget Disease of BoneenENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema NodosumlosLOSNeisseria huesos largos, y el diagnóstico se confirma por las características patológicas, como histiocitos espumosos y células gigantes de Touton (histiocitos multinucleados) enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum un fondo de fibrosisFibrosisAny pathological condition where fibrous connective tissue invades any organ, usually as a consequence of inflammation or other injury.Bronchiolitis Obliterans (CD1a no se expresa).
Sarcoma de células de Langerhans: neoplasia de alto grado que se presenta con afectación de un solo órgano o multisistémica. La presentación clínica es similar a la de la HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have “hair-like projections” visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia, pero puede distinguirse por el análisis citológico. El sarcoma de células de Langerhans se caracteriza por una citología maligna (atipia celular y mitosisMitosisA type of cell nucleus division by means of which the two daughter nuclei normally receive identical complements of the number of chromosomes of the somatic cells of the species.Cell Cycle atípicas).
Xantogranuloma juvenil: forma más frecuente de HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have “hair-like projections” visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia no HCLHCLHairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes that have “hair-like projections” visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow (causing fibrosis), and red pulp of the spleen, leading to cytopenias.Hairy Cell Leukemia. El xantogranuloma juvenil afecta predominantemente a niños y se presenta clínicamente con lesiones cutáneas solitarias o múltiples. La dermatoscopia ayuda alALAmyloidosis diagnóstico. La afección puede resolverse espontáneamente o requerir escisiones cutáneas curativas.
Enfermedad de Rosai-Dorfman: proliferación anormal y acumulación de histiocitos enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema NodosumlosLOSNeisseria ganglios linfáticos, con rara aparición de afectación extraganglionar. LosLOSNeisseria individuos pueden presentar linfadenopatía (cervical), fiebre, palidez, pérdida de peso, malestar y rinitis crónica. El diagnóstico implica el examen histopatológico (negativo para CD1a y CD207). Puede producirse una remisión espontánea. LosLOSNeisseria tratamientos incluyen terapia de soporte, administración de esteroides o interferón alfa, o quimioterapia.
Emile, J. F., Abla, O., Fraitag, S., Horne, A., Haroche, J., Donadieu, J., Requena-Caballero, L., Jordan, M. B., Abdel-Wahab, O., Allen, C. E., Charlotte, F., Diamond, E. L., Egeler, R. M., Fischer, A., Herrera, J. G., Henter, J. I., Janku, F., Merad, M., Picarsic, J., Rodriguez-Galindo, C., … Histiocyte Society. (2016). Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood, 127(22), 2672–2681. https://doi.org/10.1182/blood-2016-01-690636
Goyal, G., Young, J. R., Koster, M. J., Tobin, W. O., Vassallo, R., Ryu, J. H., Davidge-Pitts, C. J., Hurtado, M. D., Ravindran, A., Sartori Valinotti, J. C., Bennani, N. N., Shah, M. V., Rech, K. L., Go, R. S., Mayo Clinic Histiocytosis Working Group. (2019). The Mayo Clinic Histiocytosis Working Group consensus statement for the diagnosis and evaluation of adult patients with histiocytic neoplasms: Erdheim-Chester disease, Langerhans cell histiocytosis, and Rosai-Dorfman disease. Mayo Clinic Proceedings, 94(10), 2054–2071. https://doi.org/10.1016/j.mayocp.2019.02.023
Allen, C. E., Merad, M., McClain, K. L. (2018). Langerhans-cell histiocytosis. New England Journal of Medicine, 379(9), 856–868. https://doi.org/10.1056/NEJMra1607548
Jezierska, M., Stefanowicz, J., Romanowicz, G., Kosiak, W., Lange, M. (2018). Langerhans cell histiocytosis in children—a disease with many faces: recent advances in pathogenesis, diagnostic examinations and treatment. Advances in Dermatology and Allergology, 35(1), 6–17. https://doi.org/10.5114/pdia.2017.67095
Kumar, V., Abbas, A., Aster, J., Turner, J. (2021). Diseases of the white blood cells, lymph nodes, spleen and thyroid. In Robbins and Cotran Pathologic Basis of Disease (10th ed., pp. 627–628). Elsevier.
Go, R. S., et al. (2021). Histiocytic neoplasms, version 2.2021, NCCN clinical practice guidelines in oncology. JNCCN, 19(11), 1277–1303. https://doi.org/10.6004/jnccn.2021.0053
Vassallo, R., Harari, S., Tazi, A. (2017). Current understanding and management of pulmonary Langerhans cell histiocytosis. Thorax, 72(10), 937–945. https://doi.org/10.1136/thoraxjnl-2017-210125
¡Crea tu cuenta gratis o inicia una sesión para seguir leyendo!
Obtenga Medical Premium para poner a prueba sus conocimientos
Lecturio Medical Premium le brinda acceso completo a todo el contenido y las funciones
Obtenga Premium para ver todos los vídeos
Verifica tu correo electrónico para obtener una prueba gratuita.
Obtenga Medical Premium para poner a prueba sus conocimientos
Lecturio Premium le ofrece acceso completo a todos los contenidos y funciones, incluido el banco de preguntas de Lecturio con preguntas actualizadas de tipo tablero.