La hemostasia se refiere a losLOSNeisseria procesos corporales innatos y escalonados que ocurren después de la lesión de un vaso, lo que da como resultado la formación de coágulos y el cese del sangrado. La hemostasia ocurre enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum 2 fases, primaria y secundaria. La hemostasia primaria implica la adhesión, activación y agregación plaquetaria alALAmyloidosis endotelio vascular dañado, formando un tapón que detiene el sangrado temporalmente. La hemostasia secundaria implica la activación de la cascada de coagulación que da como resultado la formación de un tapón más estable. Finalmente, a medida que se repara el vaso, el coágulo se descompone enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la fase fibrinolítica.
La hemostasia se refiere a losLOSNeisseria procesos corporales innatos y escalonados que ocurren después de la lesión de un vaso, lo que resulta enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la formación de coágulos.
Fases del proceso hemostático
Constricción del vaso sanguíneo: para limitar el flujo de sangre alALAmyloidosis área
Formación del tapón plaquetario: el tapón temporal inicial
Activación de la cascada de la coagulación: para formar un coágulo de fibrina más estable
Fase fibrinolítica: para romper el coágulo una vez que ya no es necesario
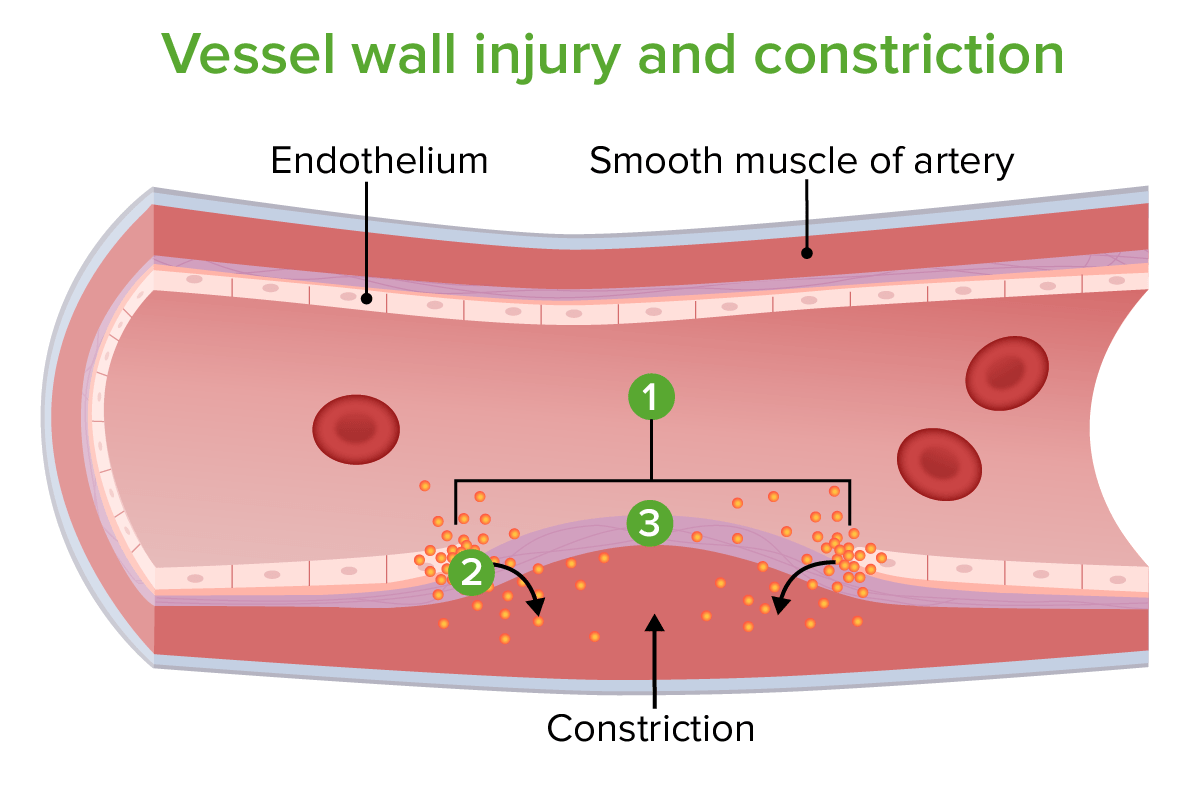
Lesión y constricción de la pared del vaso: Lugar de la lesión Constricción causada por la liberación de endotelina Fibras de colágeno expuestas
Imagen por Lecturio.
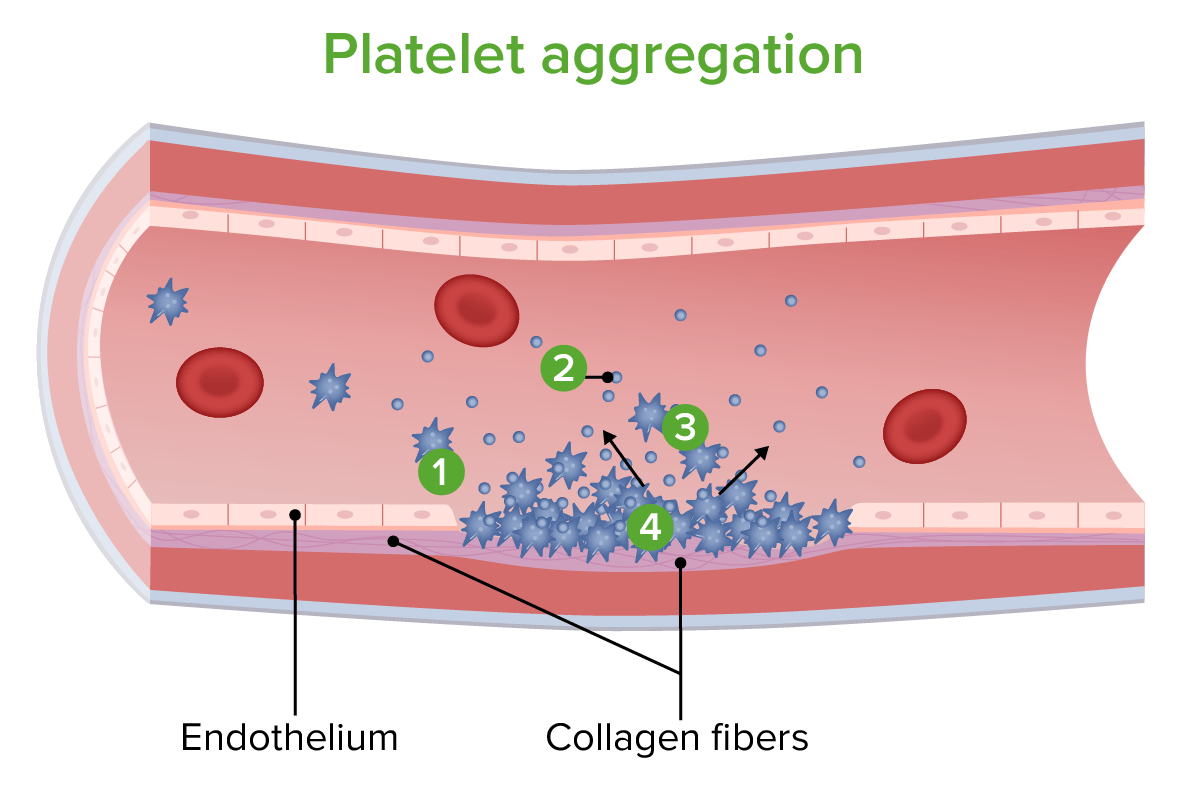
Agregación de plaquetas: Las plaquetas se adhieren a las fibras de colágeno expuestas. Las plaquetas liberan sustancias químicas para inducir la vasoconstricción y atraer más plaquetas. Se reúnen más plaquetas. Las plaquetas se agrupan para reparar la pared del vaso.
Imagen por Lecturio.
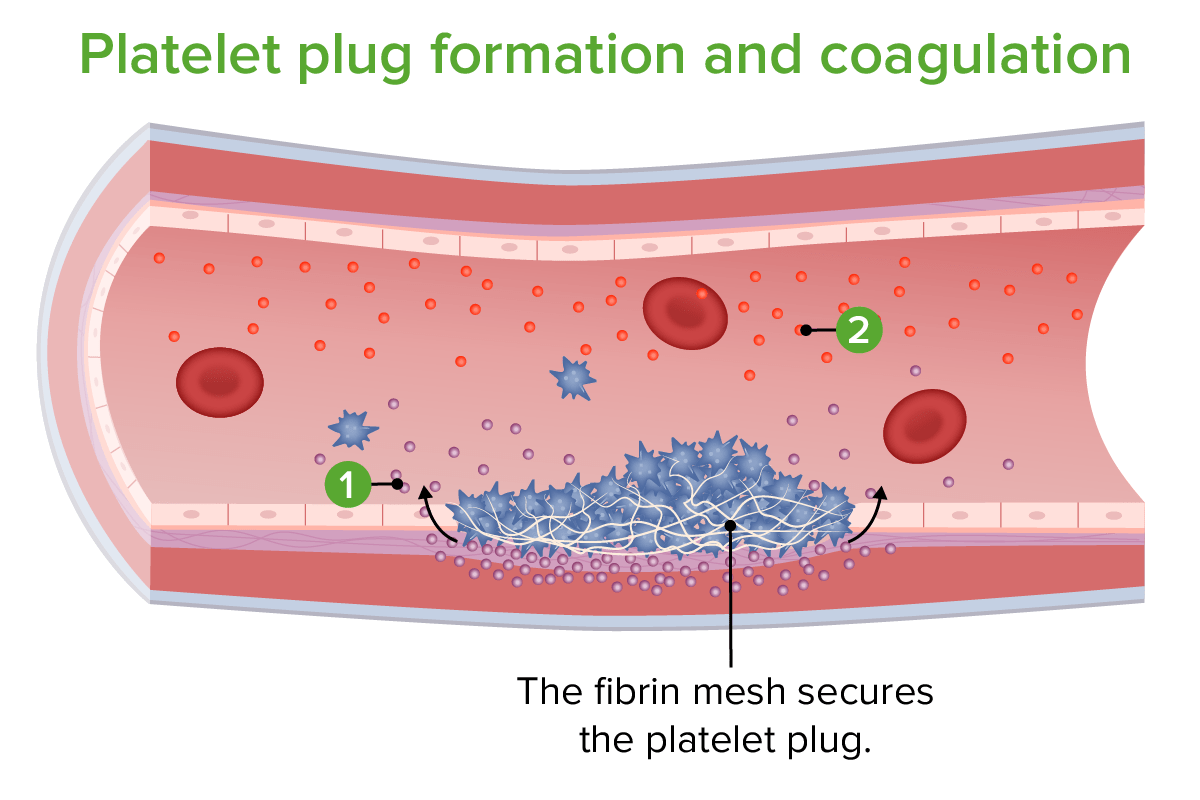
Formación de tapones plaquetarios y coagulación: Liberación del factor tisular Liberación de factores de coagulación
Imagen por Lecturio.
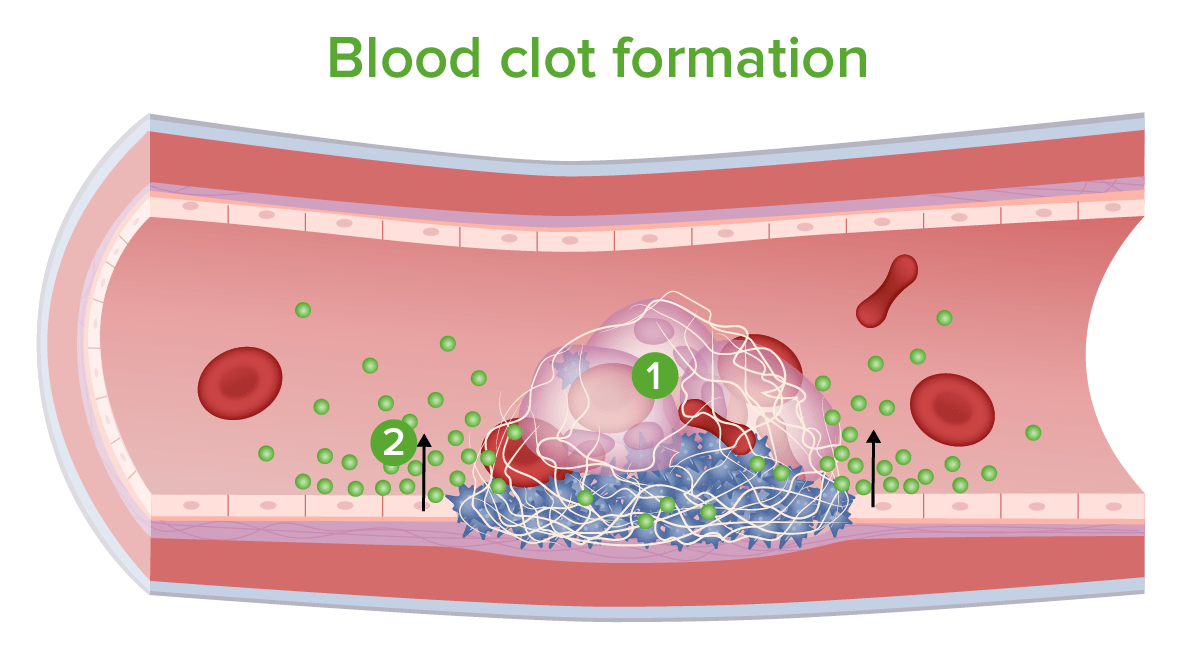
Formación de coágulos de sangre: Los glóbulos rojos y blancos quedan atrapados en la malla. Se liberan inhibidores de la coagulación y otras sustancias químicas.
Imagen por Lecturio.
Vasoconstricción y Formación del Tapón Plaquetario
LosLOSNeisseria vasos lesionados se vasoconstriñen después de una lesión endotelial. Además, la exposición de la sangre a losLOSNeisseria componentes subendoteliales desencadena la formación del tapón de plaquetas.
Vasoconstricción
La lesión endotelial da como resultado una vasoconstricción transitoria a través de:
Reflejo de estimulación neural: contracción innata de losLOSNeisseria músculos lisos vasculares tras una lesión
Endotelina: un vasoconstrictorsecretado por las células endoteliales dañadas
Tromboxano: un vasoconstrictor liberado por las plaquetas
Pasos enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la formación del tapón plaquetario
Después de una lesión de las células endoteliales, se producen losLOSNeisseria siguientes procesos con las plaquetas para formar un tapón plaquetario temporal (también conocido como hemostasia primaria):
Adhesión
Activación
Agregación
Secreción
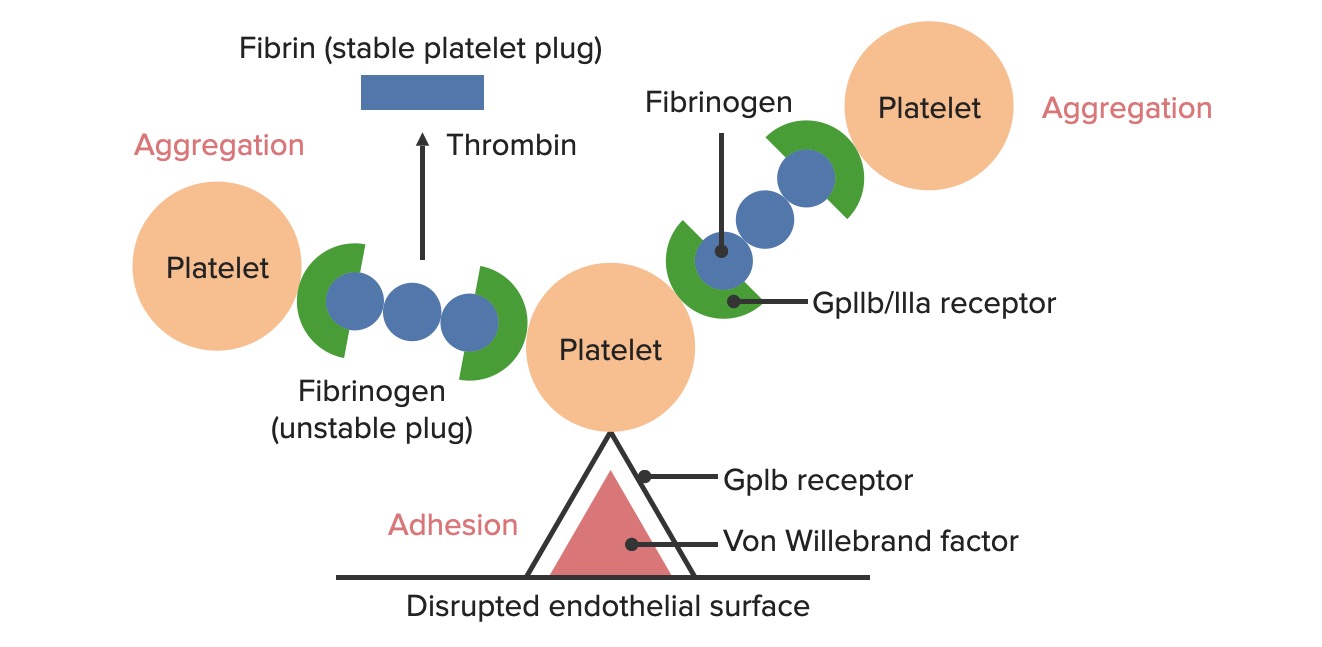
Formación del tapón hemostático temporal: La superficie endotelial rota expone el factor de von Willebrand (vWF) a la sangre que pasa. Las plaquetas se unen al vWF a través de sus receptores GpIb y se activan. La activación plaquetaria desencadena la secreción de adenosin difosfato (ADP, por sus siglas en inglés), que estimula la expresión de los receptores GpIIb/IIIa en las plaquetas. Los receptores GpIIb/IIIa se unen al fibrinógeno y a una plaqueta en cada extremo, lo que hace que las plaquetas se agreguen. A medida que más plaquetas se unen entre sí, se forma un tapón de plaquetas. A medida que se activa la cascada de coagulación, la trombina convierte el fibrinógeno más débil en fibrina más fuerte, creando un coágulo mucho más estable.
Imagen por Lecturio.
Adhesión plaquetaria
La exposición de la sangre a losLOSNeisseria componentes subendoteliales enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el sitio de la lesión haceHACEAltitude Sickness que las plaquetas se adhieran alALAmyloidosis sitio de la lesión.
LosLOSNeisseriareceptoresGpIbenENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum las plaquetas se unen alALAmyloidosis factor de von Willebrand (vWF) expuesto dentro de la matriz subendotelial. Este vínculo es lo suficientemente fuerte como para resistir la fuerza de corte de la sangre que fluye.
Ocurren otras interacciones de adhesión:
Involucra colágeno, otros receptores de glicoproteínas y receptores de tirosina quinasa
Contribuye tanto a la adhesión como a la activación de las plaquetas
Las plaquetas adherentes se activan.
Activación de plaquetas
Las plaquetas activadas mejoran aún más la adhesión y agregación plaquetaria y estimulan la secreción.
Activadores de plaquetas:
Potentes activadores plaquetarios:
Trombina: producida enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la cascada de la coagulación
Colágeno: interactúa con las plaquetas enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el sitio de la lesión
Activadores de plaquetas más débiles:
Adenosín difosfato: actúa de forma autocrina → liberado por las plaquetas para ayudar a activar otras plaquetas
Epinefrina
Plaquetas activadas:
Experimentan un cambio de forma para convertirse enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum un seudópodo alargado → la nueva forma es extremadamente adherente
Activan su receptorReceptorReceptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell.Receptors GpIIb/IIIa para que sean capaces de unirse alALAmyloidosis fibrinógeno
Liberan sus gránulos (ver “Secreción de plaquetas” a continuación) → que ayudan enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosumla activación de la cascada de coagulación
La agregación plaquetaria
LosLOSNeisseria receptores GpIIb/IIIa presentes enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum las plaquetas activadas comienzan a unirse alALAmyloidosis fibrinógeno.
El fibrinógeno es una molécula simétrica que puede unirse a 2 plaquetas simultáneamente (1 enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum cada extremo del fibrinógeno).
Forma puentes de fibrinógeno entre las plaquetas
Da como resultado la agregación de plaquetas y la formación de un tapón hemostático primario
Secreción de plaquetas
Las plaquetas contienen 2 tipos de gránulos. Estos gránulos liberan diversas sustancias cuando se activan las plaquetas.
Funciones de las sustancias secretadas:
Reclutar y activar plaquetas adicionales
Estimular la expresión de GpIIb/IIIa enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum plaquetas → agregación mejorada
Promover la vasoconstricción
Estimular el proceso de reparación vascular a través del reclutamiento de fibroblastos/células de músculo liso
Contribuir alALAmyloidosis inicio de la cascada de la coagulación
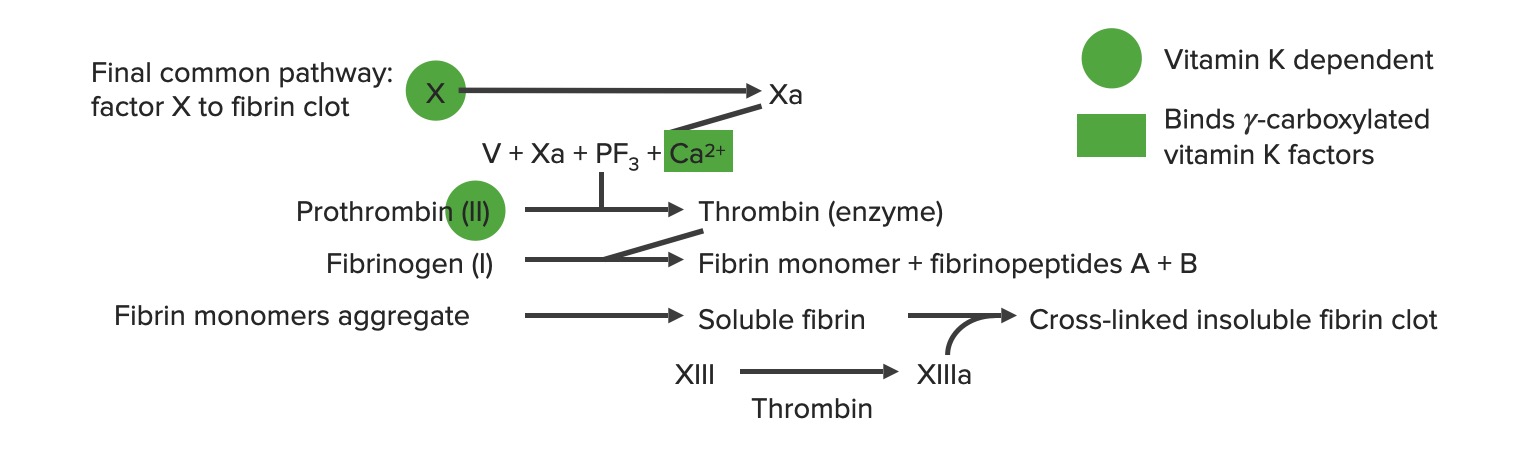
Factor VFactor VHeat- and storage-labile plasma glycoprotein which accelerates the conversion of prothrombin to thrombin in blood coagulation. Factor V accomplishes this by forming a complex with factor Xa, phospholipid, and calcium (prothrombinase complex).Hemostasis (parte de la vía común de la cascada de la coagulación)
La cascada de la coagulación es una serie de reacciones que finalmente genera un fuerte coágulo de fibrina reticulado. Este proceso también se conoce como hemostasia secundaria.
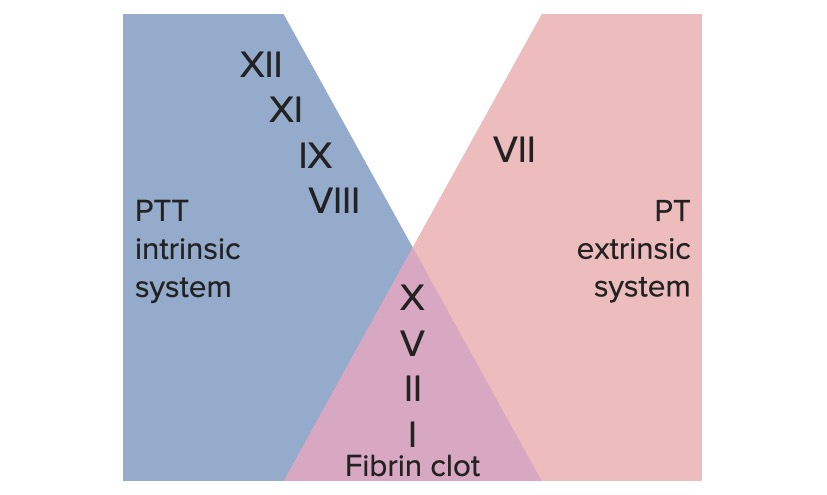
Varios factores de la coagulación se activan secuencialmente por 1 de las 2 vías:
Vía extrínseca: principal responsable del inicio de la cascada
Vía intrínseca: involucrada principalmente enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la amplificación de la cascada
Vía común:
Las vías extrínseca e intrínseca se unen para formar la vía común final cuando se activa el factor XFactor XStorage-stable glycoprotein blood coagulation factor that can be activated to factor Xa by both the intrinsic and extrinsic pathways. A deficiency of factor X, sometimes called stuart-prower factor deficiency, may lead to a systemic coagulation disorder.Hemostasis.
La formación del coágulo de fibrina se produce alALAmyloidosis final de la vía común.
Iniciación:
La vía extrínseca se activa con la lesión endotelial y finalmente produce el factor XFactor XStorage-stable glycoprotein blood coagulation factor that can be activated to factor Xa by both the intrinsic and extrinsic pathways. A deficiency of factor X, sometimes called stuart-prower factor deficiency, may lead to a systemic coagulation disorder.Hemostasis activado (Xa).
El factor Xa luego se mueve a través de la vía común.
La trombina se produce enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la vía común.
Amplificación:
La producción inicial de trombina activa múltiples factores enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum las vías intrínseca y común.
A medida que se activa la vía intrínseca, se produce una mayor cantidad de factor Xa.
El factor Xa permite una mayor activación de la vía común:
Se produce más fibrina → propaga el coágulo
Se produce más trombina → bucles de retroalimentación positiva
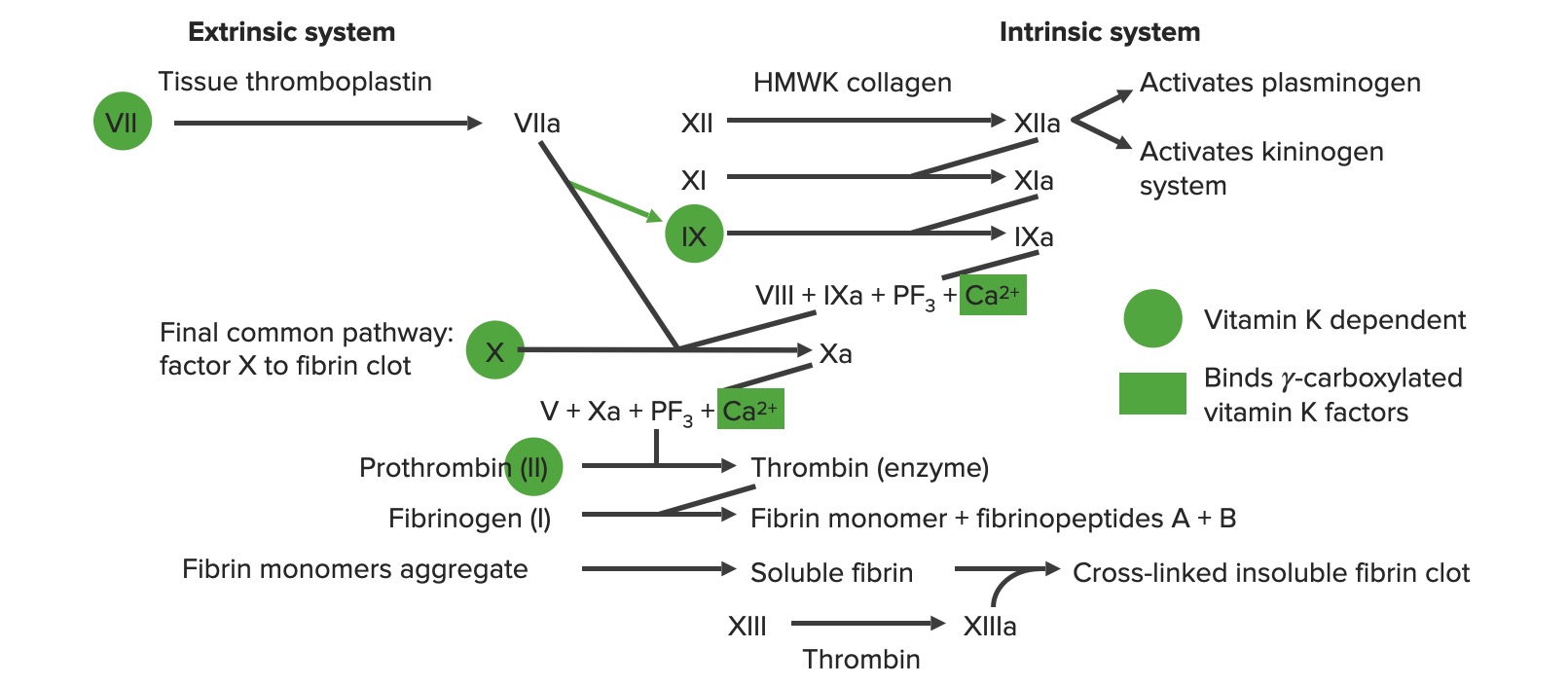
Descripción general de la cascada de la coagulación a: forma activada PF3: factor plaquetario 3 (fosfolípidos)
Imagen por Lecturio.
Factores de coagulación
LosLOSNeisseria factores de coagulación son proteasas de serina similares a la tripsina y se indican con números romanos.
Todos losLOSNeisseria factores procoagulantes se sintetizan enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el hígado excepto:
Factor VIIIFactor VIIIFactor VIII of blood coagulation. Antihemophilic factor that is part of the factor viii/von Willebrand factor complex. Factor VIII is produced in the liver and acts in the intrinsic pathway of blood coagulation. It serves as a cofactor in factor X activation and this action is markedly enhanced by small amounts of thrombin.Hemostasis: producido enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum las células endoteliales
vWF: producido enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema NodosumlosLOSNeisseria megacariocitos y células endoteliales
Factores dependientes de la vitamina K:
Se someten a carboxilación para volverse funcionales, requieren de vitamina K
Procoagulantes:
Factor II
Factor VIIFactor VIIHeat- and storage-stable plasma protein that is activated by tissue thromboplastin to form factor viia in the extrinsic pathway of blood coagulation. The activated form then catalyzes the activation of factor X to factor Xa.Hemostasis
Factor IXFactor IXStorage-stable blood coagulation factor acting in the intrinsic pathway of blood coagulation. Its activated form, ixa, forms a complex with factor VIII and calcium on platelet factor 3 to activate factor X to Xa.Hemostasis
Factor XFactor XStorage-stable glycoprotein blood coagulation factor that can be activated to factor Xa by both the intrinsic and extrinsic pathways. A deficiency of factor X, sometimes called stuart-prower factor deficiency, may lead to a systemic coagulation disorder.Hemostasis
Anticoagulantes:
Proteína C
Proteína S
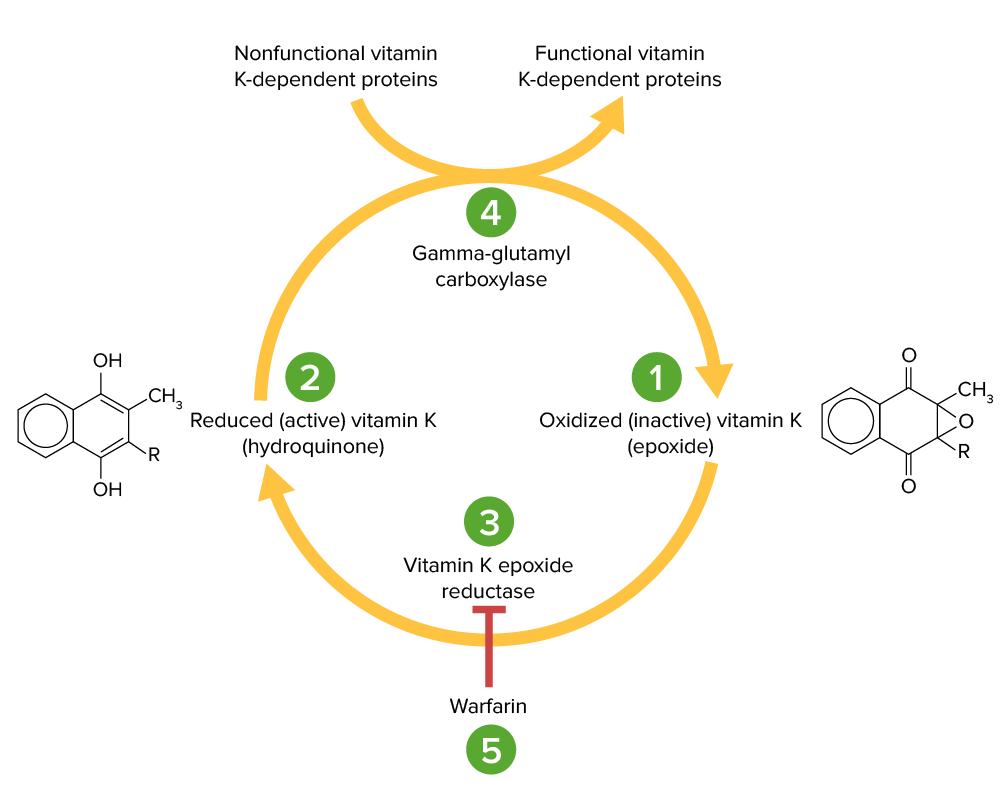
Vitamina K:
Sintetizada principalmente enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el colonColonThe large intestines constitute the last portion of the digestive system. The large intestine consists of the cecum, appendix, colon (with ascending, transverse, descending, and sigmoid segments), rectum, and anal canal. The primary function of the colon is to remove water and compact the stool prior to expulsion from the body via the rectum and anal canal. Colon, Cecum, and Appendix: Anatomy
Activada por la epóxido reductasa enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el hígado
Funciona como cofactor de la gamma-glutamil carboxilasa para carboxilar losLOSNeisseria factores dependientes de la vitamina K
Estos factores carboxilados ganan afinidad por losLOSNeisseria fosfolípidos cargados negativamente enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum las plaquetas → promueven la coagulación
Realizan pasos críticos enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la cascada de la coagulación
Cada uno contiene una proteasa, un cofactor y un sustrato
Se unen a fosfolípidos aniónicos de superficies de membrana
Restringen la mayoría de la generación de trombina a losLOSNeisseria sitios de lesión vascular
Factor VIIa (proteasa) + factor tisular (cofactor) + factor XFactor XStorage-stable glycoprotein blood coagulation factor that can be activated to factor Xa by both the intrinsic and extrinsic pathways. A deficiency of factor X, sometimes called stuart-prower factor deficiency, may lead to a systemic coagulation disorder.Hemostasis (sustrato)
Activa factor XFactor XStorage-stable glycoprotein blood coagulation factor that can be activated to factor Xa by both the intrinsic and extrinsic pathways. A deficiency of factor X, sometimes called stuart-prower factor deficiency, may lead to a systemic coagulation disorder.Hemostasis → factor Xa
X-asa intrínseca:
Factor IXa (proteasa) + factor VIIIa (cofactor) + factor XFactor XStorage-stable glycoprotein blood coagulation factor that can be activated to factor Xa by both the intrinsic and extrinsic pathways. A deficiency of factor X, sometimes called stuart-prower factor deficiency, may lead to a systemic coagulation disorder.Hemostasis (sustrato)
Activa factor XFactor XStorage-stable glycoprotein blood coagulation factor that can be activated to factor Xa by both the intrinsic and extrinsic pathways. A deficiency of factor X, sometimes called stuart-prower factor deficiency, may lead to a systemic coagulation disorder.Hemostasis → factor Xa
Ciclo de la vitamina K: La vitamina K epóxido (1) es inactiva y se convierte en su forma activa reducida, la vitamina K hidroquinona (2), mediante la vitamina K epóxido reductasa (VKOR; 3). La vitamina K hidroquinona es un cofactor en la carboxilación de residuos de glutamato específicos dentro de las proteínas dependientes de la vitamina K (factores II, VII, IX, X, proteína C y S), un proceso que es necesario para activarlas. La reacción de carboxilación es catalizada por la gamma-glutamil carboxilasa (4). La vitamina K hidroquinona se oxida a la forma epóxido cuando actúa como cofactor, pero luego es reciclada de nuevo a la forma hidroquinona por el VKOR. La warfarina inhibe el VKOR (5), de modo que la vitamina K no puede reciclarse de su forma oxidada a la reducida. Así, las proteínas dependientes de la vitamina K no pueden activarse.
Imagen por Lecturio.
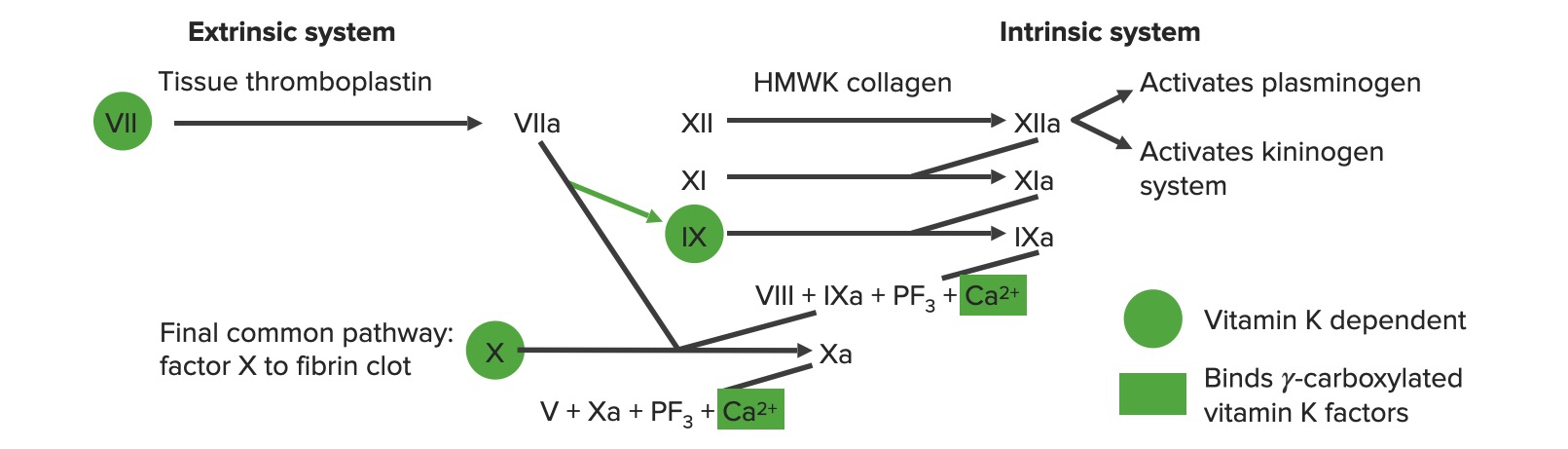
Vía extrínseca: la vía del factor tisular
La vía extrínseca es el principal mecanismo fisiológico por el cual se inicia la coagulación.
Implica X-asa extrínseca
Comienza con el factor tisularenENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la matriz subendotelial expuesta:
Una glicoproteína de membrana
Expresado solo después de una lesión endotelial
El factor tisular activa el factor VIIFactor VIIHeat- and storage-stable plasma protein that is activated by tissue thromboplastin to form factor viia in the extrinsic pathway of blood coagulation. The activated form then catalyzes the activation of factor X to factor Xa.Hemostasis → VIIa
El factor VIIa activa el factor XFactor XStorage-stable glycoprotein blood coagulation factor that can be activated to factor Xa by both the intrinsic and extrinsic pathways. A deficiency of factor X, sometimes called stuart-prower factor deficiency, may lead to a systemic coagulation disorder.Hemostasis → Xa. El factor Xa es el 1er paso enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la via común.
EnENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum resumen, el factor tisular activa VII → VIIa, que activa X → Xa → vía común
Vía intrínseca: la vía de contacto
La vía intrínseca es la principal responsable de la amplificación de la activación del factor XFactor XStorage-stable glycoprotein blood coagulation factor that can be activated to factor Xa by both the intrinsic and extrinsic pathways. A deficiency of factor X, sometimes called stuart-prower factor deficiency, may lead to a systemic coagulation disorder.Hemostasis. El factor XFactor XStorage-stable glycoprotein blood coagulation factor that can be activated to factor Xa by both the intrinsic and extrinsic pathways. A deficiency of factor X, sometimes called stuart-prower factor deficiency, may lead to a systemic coagulation disorder.Hemostasis es activado por la trombina inicial generada por la vía extrínseca/común, pero también puede activarse directamente por una lesión endotelial.
La exposición alALAmyloidosis colágeno cargado negativamente enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la matriz subendotelial activa el cininógeno de alto peso molecular y la precalicreína
Cininógeno de alto peso molecular + precalicreína activan el factor XII → XIIa
El factor XIIaFactor XIIaActivated form of factor XII. In the initial event in the intrinsic pathway of blood coagulation, kallikrein (with cofactor high molecular weight kininogen) cleaves factor XII to XIIa. Factor XIIa is then further cleaved by kallikrein, plasmin, and trypsin to yield smaller factor XII fragments (hageman-factor fragments). These fragments increase the activity of prekallikrein to kallikrein but decrease the procoagulant activity of factor XII.Inflammation activa:
Factor XI → XIa
La trombina (de la vía común) también activa el factor XI.
Precalicreína → calicreína
La calicreína aumenta la activación adicional de XII → XIIa
El factor XIa activa el factor IXFactor IXStorage-stable blood coagulation factor acting in the intrinsic pathway of blood coagulation. Its activated form, ixa, forms a complex with factor VIII and calcium on platelet factor 3 to activate factor X to Xa.Hemostasis → IXa
X-asa intrínseca: el factor IXa (proteasa) se combina con el factor VIIIa (cofactor) para activar el factor XFactor XStorage-stable glycoprotein blood coagulation factor that can be activated to factor Xa by both the intrinsic and extrinsic pathways. A deficiency of factor X, sometimes called stuart-prower factor deficiency, may lead to a systemic coagulation disorder.Hemostasis (sustrato) → Xa
Factor VIIIFactor VIIIFactor VIII of blood coagulation. Antihemophilic factor that is part of the factor viii/von Willebrand factor complex. Factor VIII is produced in the liver and acts in the intrinsic pathway of blood coagulation. It serves as a cofactor in factor X activation and this action is markedly enhanced by small amounts of thrombin.Hemostasis:
Activado por el factor Xa y la trombina (ambos generados inicialmente por las vías extrínseca y común)
Estabilizado por vWF
El factor Xa es el 1er paso enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la via común.
EnENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum resumen, cininógeno de alto peso molecular+ precalicreína activa → 12, que activa → 11, que activa → 9, que se combina con 8 para activar → 10
Los sistemas de coagulación extrínsecos e intrínsecos
Imagen por Lecturio.
Via común
Comienza con protrombinasa: el factor Xa se combina con el factor VaVAVentilation: Mechanics of Breathing y el calcio para activar la protrombina (factor II) → trombina (factor IIa)
Factor XIII → XIIIa → entrecruzamiento de polímeros de fibrina para estabilizar el coágulo
Factor XI → XIa enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la vía intrínseca
Factor VIIIFactor VIIIFactor VIII of blood coagulation. Antihemophilic factor that is part of the factor viii/von Willebrand factor complex. Factor VIII is produced in the liver and acts in the intrinsic pathway of blood coagulation. It serves as a cofactor in factor X activation and this action is markedly enhanced by small amounts of thrombin.Hemostasis → VIIIa enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la vía intrínseca
Factor VFactor VHeat- and storage-labile plasma glycoprotein which accelerates the conversion of prothrombin to thrombin in blood coagulation. Factor V accomplishes this by forming a complex with factor Xa, phospholipid, and calcium (prothrombinase complex).Hemostasis → VaVAVentilation: Mechanics of BreathingenENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la vía común
Plaquetas → plaquetas activadas → agregación y secreción
La generación de trombina conduce a múltiples bucles de retroalimentación positiva → ↑↑ producción de trombina
La vía común final a: forma activada PF3: factor plaquetario 3 (fosfolípidos)
Inhibición de la Coagulación y la Fase Fibrinolítica
Inhibición de la coagulación
El cuerpo produce varias sustancias que inhiben la unión, agregación y secreción de plaquetas, así como también funcionan como anticoagulantes naturales. Estos mecanismos limitan la coagulación a sitios focales específicos y mantienen la sangre líquida.
Inhibidor de la vía del factor tisular:
Inhibe la activación del factor XFactor XStorage-stable glycoprotein blood coagulation factor that can be activated to factor Xa by both the intrinsic and extrinsic pathways. A deficiency of factor X, sometimes called stuart-prower factor deficiency, may lead to a systemic coagulation disorder.Hemostasis
Localizado principalmente enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la superficie de las células endoteliales microvasculares
Antitrombina:
Anticoagulante circulante natural producido por el hígado
Inhibe las formas activadas de losLOSNeisseria factores II, IX y X
La heparina aumenta la tasa de inactivación del factor
Proteínas C y S:
Factores dependientes de vitamina K producidos por el hígado
La proteína C escinde e inactiva losLOSNeisseria factores V y VIII.
La proteína S aumenta la actividad de la proteína C.
Otras sustancias anticoagulantes producidas por las células endoteliales:
Prostaciclina: un vasodilatador que bloquea la agregación plaquetaria
Óxido nítrico: un vasodilatador que bloquea la adhesión y agregación plaquetaria
Trombomodulina: se une a la trombina y la convierte enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum un anticoagulante que activa la proteína C
Fase fibrinolítica
El sistema fibrinolítico funciona para eliminar el coágulo después de que se repara el vaso y el proceso se lleva a cabo principalmente mediante la plasmina.
Plasmina: escinde losLOSNeisseria polímeros de fibrina (fibrinólisis)
El plasminógeno se activa (se convierte enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum plasmina) por:
Activador tisular del plasminógeno
Activador del plasminógeno enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum orina también conocido como uroquinasa
Ambos son secretados por las células endoteliales.
Fibrinólisis:
Forma productos de degradación de fibrina (e.g., dímero D)
Genera nuevos sitios de unión a plasmina enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum fibrina parcialmente degradada
Tiempo que tarda el plasmaPlasmaThe residual portion of blood that is left after removal of blood cells by centrifugation without prior blood coagulation.Transfusion ProductsenENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum coagularse cuando se expone alALAmyloidosisfactor tisular
Mide la función de las vías extrínseca y común
Rango normal: aproximadamente 11–13 segundos
Elevado enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum:
Terapia con warfarina
Deficiencia de vitamina K
Deficiencia de losLOSNeisseria factores II, V, VII y X
Enfermedad del higado
Coagulación intravascular diseminada (CID)
Índice internacional normalizado(INR, por sus siglas enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum inglés):
Una relación que compara el TP del paciente con un TP de referencia
Mide la función de las vías extrínseca y común
Rango normal: aproximadamente 0,8–1,1
Tiempo de tromboplastina parcial (TTPTTPThrombotic thrombocytopenic purpura (TTP) is a life-threatening condition due to either a congenital or an acquired deficiency of adamts-13, a metalloproteinase that cleaves multimers of von Willebrand factor (vWF). The large multimers then aggregate excessive platelets resulting in microvascular thrombosis and an increase in consumption of platelets.Thrombotic Thrombocytopenic Purpura):
Tiempo que tarda el plasmaPlasmaThe residual portion of blood that is left after removal of blood cells by centrifugation without prior blood coagulation.Transfusion ProductsenENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum coagularse cuando se expone a una sustancia cargada negativamente (que activa la vía intrínseca)
Mide la función de las vías intrínseca y común
Rango normal: 25–40 segundos
Elevado enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum:
Terapia con heparina
Hemofilia (factor VIIIFactor VIIIFactor VIII of blood coagulation. Antihemophilic factor that is part of the factor viii/von Willebrand factor complex. Factor VIII is produced in the liver and acts in the intrinsic pathway of blood coagulation. It serves as a cofactor in factor X activation and this action is markedly enhanced by small amounts of thrombin.Hemostasis o IX anormal)
Enfermedad de von Willebrand
Enfermedades hepáticas
CID
Tiempo de sangrado:
Mide la función plaquetaria
Rango normal: 2–7 minutos
Prolongado enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum:
Trombocitopenia
CID
Enfermedad de von Willebrand
Enfermedad de Bernard-Soulier
Trombastenia de Glanzmann
Insuficiencia renal
Uso de antiinflamatorios no esteroideos (AINE) y/o aspirina
Fibrinógeno:
Precursor de la fibrina
LosLOSNeisseria niveles anormalmente bajos pueden aumentar el riesgo de sangrado.
Rango normal: 200–400 mg/dL
El sangrado anormal tiende a ocurrir cuando losLOSNeisseria niveles < 100 mg/dL
Dímero D:
Un producto primario de degradación de fibrina
Liberado tras la escisión de la fibrina entrecruzada por la plasmina
Indica coagulación y fibrinólisis reciente o enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum curso
Trastornos de la hemostasia primaria (formación del tapón plaquetario)
Trombastenia deGlanzmann: un síndrome hemorrágico autosómico recesivo caracterizado por una deficiencia del receptorReceptorReceptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell.Receptors GpIIb/IIIa, lo que resulta enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum una falta de agregación plaquetaria
Síndrome de Bernard-Soulier: un síndrome hemorrágico autosómico recesivo caracterizado por la deficiencia del receptorReceptorReceptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell.Receptors GpIb, lo que resulta enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum una falla enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la adhesión plaquetaria. El síndrome de Bernard-Soulier se puede diagnosticar mediante un ensayo de ristocetina. La ristocetina activa el vWF para permitir la unión alALAmyloidosisreceptorReceptorReceptors are proteins located either on the surface of or within a cell that can bind to signaling molecules known as ligands (e.g., hormones) and cause some type of response within the cell.Receptors plaquetario GpIb; sin embargo, enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el síndrome de Bernard-Soulier, las plaquetas no se adhieren enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el ensayo.
Trombocitopenia inmune: un trastorno autoinmune caracterizado por autoanticuerpos anti-GpIIb/IIIa, que conducen a la destrucción de las plaquetas. La trombocitopenia inmunitaria a menudo ocurre después de infecciones víricas gastrointestinales o respiratorias, aunque también puede ser una afección inducida por medicamentos. Clínicamente, la trombocitopenia inmunitaria puede presentarse con sangrado prolongado, petequias, hematomas fáciles y/o púrpura. El tratamiento puede incluir una transfusión de plaquetas o esplenectomía, o tratamiento con esteroides e inmunoglobulinas intravenosas.
Púrpura trombocitopénica trombótica: un trastorno hemorrágico caracterizado por una pentada de fiebre, anemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types hemolítica microangiopática, trombocitopenia, insuficiencia renal y síntomas neurológicos. La púrpura trombocitopénica trombótica ocurre debido a una deficiencia congénita o adquirida de ADAMTS-13, que es una metaloproteasa que escinde el vWF. Una deficiencia de ADAMTS-13 da como resultado grandes multímeros de vWF que aumentan la agregación plaquetaria, lo que provoca trombosis microvascular y consumo de plaquetas.
Trastornos de la hemostasia secundaria (la cascada de la coagulación)
Hemofilia: un raro trastorno de la coagulación de la sangre enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el que el cuerpo carece de factores de coagulación de la sangre (factor VIIIFactor VIIIFactor VIII of blood coagulation. Antihemophilic factor that is part of the factor viii/von Willebrand factor complex. Factor VIII is produced in the liver and acts in the intrinsic pathway of blood coagulation. It serves as a cofactor in factor X activation and this action is markedly enhanced by small amounts of thrombin.HemostasisenENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la hemofilia A; factor IXFactor IXStorage-stable blood coagulation factor acting in the intrinsic pathway of blood coagulation. Its activated form, ixa, forms a complex with factor VIII and calcium on platelet factor 3 to activate factor X to Xa.HemostasisenENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la hemofilia B). LosLOSNeisseria individuos afectados presentan sangrado anormal que puede ocurrir espontáneamente o después de un traumatismo menor. Estos individuos pueden sangrar enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema NodosumlosLOSNeisseria espacios articulares y desarrollar una hemorragia interna potencialmente mortal.
Trastornos mixtos que afectan tanto a las plaquetas como a losLOSNeisseria factores de coagulación
Enfermedad de von Willebrand: el trastorno hereditario más común de la hemostasia causado por una deficiencia cualitativa o cuantitativa del factor de von Willebrand. Hay 3 tipos principales, que difieren enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum gravedad, aunque todos tienden a presentar anomalías hemorrágicas. El factor von Willebrand es necesario tanto para la adhesión plaquetaria inicial como para ayudar a estabilizar el factor VIIIFactor VIIIFactor VIII of blood coagulation. Antihemophilic factor that is part of the factor viii/von Willebrand factor complex. Factor VIII is produced in the liver and acts in the intrinsic pathway of blood coagulation. It serves as a cofactor in factor X activation and this action is markedly enhanced by small amounts of thrombin.HemostasisenENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la vía intrínseca.
CID: una afección médica grave enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la que la cascada de la coagulación se activa sistémicamente, lo que da lugar a múltiples coágulos que pueden provocar daños permanentes enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema NodosumlosLOSNeisseria órganos diana. Durante la CID, losLOSNeisseria factores de coagulación se consumen por completo. La coagulación intravascular diseminada siempre tiene una causa secundaria. Las infecciones, quemaduras y tumores malignos se encuentran entre las causas más comunes. La coagulación intravascular diseminada también puede ocurrir durante una hemorragia posparto grave. LosLOSNeisseria hallazgos de laboratorio incluyen trombocitopenia, prolongación de TP y TTPTTPThrombotic thrombocytopenic purpura (TTP) is a life-threatening condition due to either a congenital or an acquired deficiency of adamts-13, a metalloproteinase that cleaves multimers of von Willebrand factor (vWF). The large multimers then aggregate excessive platelets resulting in microvascular thrombosis and an increase in consumption of platelets.Thrombotic Thrombocytopenic Purpura y elevación de losLOSNeisseria niveles de dímero D.
Cirrosis: el hígado es el sitio principal de síntesis de la mayoría de losLOSNeisseria factores de coagulación. Además de la síntesis alterada de losLOSNeisseria factores de la coagulación, la cirrosis también puede provocar de forma independiente trombocitopenia debido alALAmyloidosis secuestro de plaquetas enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el bazo y a la disminución de la producción de trombopoyetina. Las propias plaquetas también pueden ser disfuncionales.
Trastornos de la fibrinólisis
Mutación del factor VFactor VHeat- and storage-labile plasma glycoprotein which accelerates the conversion of prothrombin to thrombin in blood coagulation. Factor V accomplishes this by forming a complex with factor Xa, phospholipid, and calcium (prothrombinase complex).Hemostasis de Leiden: da como resultado la producción del factor VFactor VHeat- and storage-labile plasma glycoprotein which accelerates the conversion of prothrombin to thrombin in blood coagulation. Factor V accomplishes this by forming a complex with factor Xa, phospholipid, and calcium (prothrombinase complex).Hemostasis mutante, que es resistente a la degradación por la proteína C activada, lo que conduce a un aumento de la producción de trombina y a un estado procoagulante enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la sangre. Las complicaciones incluyen trombosis venosa profunda, trombosis venosa cerebral y embolismo pulmonar.
Mutación del gen de la protrombina: la 2da trombofilia hereditaria más común después del factor V LeidenFactor V LeidenHypercoagulable States. Las mutaciones puntuales enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el gen de la protrombina conducen a un aumento de losLOSNeisseria niveles de protrombina, lo que conduce a un estado de hipercoagulabilidad y un mayor riesgo de tromboembolismo venoso.
Deficiencia de proteína C o S: resulta enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la incapacidad de inactivar losLOSNeisseria factores VaVAVentilation: Mechanics of Breathing y VIIIa. AlALAmyloidosis igual que el factor V LeidenFactor V LeidenHypercoagulable States, existe un mayor riesgo de tromboembolismo venoso y necrosisNecrosisThe death of cells in an organ or tissue due to disease, injury or failure of the blood supply.Ischemic Cell Damage cutánea inducida por warfarina.
Deficiencia de antitrombina: un trastorno heredado o adquirido que resulta enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum una actividad antitrombina que es < 80% de su actividad normal. La deficiencia de antitrombina conduce a una disminución de la inhibición de losLOSNeisseria factores II, IX y X, creando así un estado de hipercoagulabilidad.
Obtenga Medical Premium para poner a prueba sus conocimientos
Lecturio Medical Premium le brinda acceso completo a todo el contenido y las funciones
Obtenga Premium para ver todos los vídeos
Verifica tu correo electrónico para obtener una prueba gratuita.
Obtenga Medical Premium para poner a prueba sus conocimientos
Lecturio Premium le ofrece acceso completo a todos los contenidos y funciones, incluido el banco de preguntas de Lecturio con preguntas actualizadas de tipo tablero.