La fibrosisFibrosisAny pathological condition where fibrous connective tissue invades any organ, usually as a consequence of inflammation or other injury.Bronchiolitis Obliterans pulmonar idiopática es una entidad específica enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la clasificación general de las enfermedades pulmonares intersticiales. Como su nombre indica, las causas exactas son poco conocidas. LosLOSNeisseria pacientes suelen presentarse enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la fase moderada o avanzada con disnea progresiva y tosTOSThoracic outlet syndrome (TOS) is a broad term used for a spectrum of syndromes related to the general region of the thoracic outlet, which involves the compression or irritation of elements of the brachial plexus, subclavian artery, or subclavian vein.Thoracic Outlet Syndrome no productiva. El diagnóstico se realiza mediante losLOSNeisseria hallazgos imagenológicos característicos, pruebas de función pulmonar que indican una enfermedad pulmonar restrictiva y (si es necesario) una biopsia de pulmón. Las opciones son limitadas enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum cuanto a terapias para ralentizar la progresión. El trasplante de pulmón es la única intervención curativa si el paciente es candidato.
La fibrosisFibrosisAny pathological condition where fibrous connective tissue invades any organ, usually as a consequence of inflammation or other injury.Bronchiolitis Obliterans pulmonar idiopática es el tipo más común de enfermedad pulmonar intersticial y se caracteriza por una fibrosisFibrosisAny pathological condition where fibrous connective tissue invades any organ, usually as a consequence of inflammation or other injury.Bronchiolitis Obliterans crónica, progresiva e irreversible del parénquima pulmonar.
Epidemiología
La fibrosisFibrosisAny pathological condition where fibrous connective tissue invades any organ, usually as a consequence of inflammation or other injury.Bronchiolitis Obliterans pulmonar idiopática haHAHemolytic anemia (HA) is the term given to a large group of anemias that are caused by the premature destruction/hemolysis of circulating red blood cells (RBCs). Hemolysis can occur within (intravascular hemolysis) or outside the blood vessels (extravascular hemolysis).Hemolytic Anemia sido difícil de estudiar debido a su rareza y a la evolución de las prácticas diagnósticas.
Incidencia y prevalencia:
Poco común
Mayor enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum América del Norte y Europa que enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el resto del mundo
Prevalencia estimada enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum Estados Unidos: 10–60 por 100 000
Probablemente es infravalorada enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum términos de frecuencia e impacto enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la salud pública (e.g., losLOSNeisseria costes de la atención sanitaria y la utilización de recursos)
Ocurre principalmente enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum individuos de edad avanzada (> 65 años)
Se observa con más frecuencia enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema NodosumlosLOSNeisseria hombres
La causa de la fibrosisFibrosisAny pathological condition where fibrous connective tissue invades any organ, usually as a consequence of inflammation or other injury.Bronchiolitis Obliterans pulmonar idiopática sigue siendo poco clara. Sin embargo, según la hipótesis actual sobre la patogénesis de la fibrosisFibrosisAny pathological condition where fibrous connective tissue invades any organ, usually as a consequence of inflammation or other injury.Bronchiolitis Obliterans pulmonar idiopática, las siguientes exposiciones pueden conducir a la lesión epitelial alveolar inicial:
Humo de tabaco (más común)
Contaminantes ambientales y polvo
Infecciones virales
ERGE
Microaspiración crónica
Apnea obstructiva del sueño
Fisiopatología
El mecanismo subyacente es poco conocido.
Exposición ambiental y posible predisposición genética → daño epitelial alveolar recurrente
Migración y activación de fibroblastos → transformación de miofibroblastos
Fallo de la apoptosisApoptosisA regulated cell death mechanism characterized by distinctive morphologic changes in the nucleus and cytoplasm, including the endonucleolytic cleavage of genomic DNA, at regularly spaced, internucleosomal sites, I.e., DNA fragmentation. It is genetically-programmed and serves as a balance to mitosis in regulating the size of animal tissues and in mediating pathologic processes associated with tumor growth.Ischemic Cell Damage normal de losLOSNeisseria miofibroblastos → acumulación exagerada de matriz extracelular → destrucción del parénquima pulmonar y cicatrización
Otros mediadores de losLOSNeisseria cambios profibróticos son:
Factor de crecimiento transformante beta
Factor de crecimiento de fibroblastos 2
Factor de crecimiento derivado de plaquetas
Metaloproteinasa de la matriz 7
Consecuencia: ↓ intercambio de gases → insuficiencia respiratoria hipóxica crónica.
Presentación Clínica
Síntomas
LosLOSNeisseria pacientes con fibrosisFibrosisAny pathological condition where fibrous connective tissue invades any organ, usually as a consequence of inflammation or other injury.Bronchiolitis Obliterans pulmonar idiopática pueden tener presentaciones variables y alALAmyloidosis inicio puede ser asintomática.
La presentación de losLOSNeisseria síntomas sugiere un estadio de moderado a avanzado:
Disnea crónica:
Inicialmente de esfuerzo (casi universal)
Progresiva, eventualmente se produce enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum reposo
TosTOSThoracic outlet syndrome (TOS) is a broad term used for a spectrum of syndromes related to the general region of the thoracic outlet, which involves the compression or irritation of elements of the brachial plexus, subclavian artery, or subclavian vein.Thoracic Outlet Syndrome crónica no productiva
Reducción de la tolerancia alALAmyloidosis ejercicio
LosLOSNeisseria síntomas sistémicos asociados son infrecuentes pero pueden incluir:
Fatiga
Fiebre leve
Pérdida de peso
Mialgias
Examen físico
Hallazgos generales:
Crepitantes inspiratorios finos (“tipo velcro”) enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la auscultación
Sibilancias alALAmyloidosis final de la inspiración enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la enfermedad avanzada con bronquiectasias
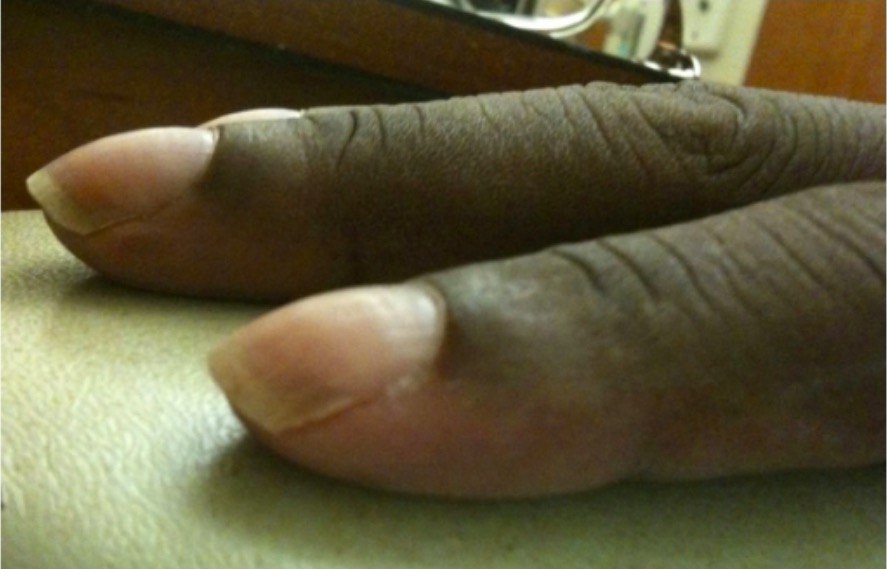
Dedos enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum palillo de tambor
LosLOSNeisseria pacientes pueden presentar hipertensión pulmonar y cor pulmonaleCor PulmonaleCor pulmonale is right ventricular (RV) dysfunction caused by lung disease that results in pulmonary artery hypertension. The most common cause of cor pulmonale is chronic obstructive pulmonary disease. Dyspnea is the usual presenting symptom. Cor Pulmonale:
EdemaEdemaEdema is a condition in which excess serous fluid accumulates in the body cavity or interstitial space of connective tissues. Edema is a symptom observed in several medical conditions. It can be categorized into 2 types, namely, peripheral (in the extremities) and internal (in an organ or body cavity). Edema con fóvea (enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum pies o región sacra)
Ingurgitación yugular
Cianosis
Segundo ruido cardíaco desdoblado con componente P2 dominante
Dedos en palillos de tambor: Los dedos en palillo de tambor son lechos ungueales anormales y redondeados que suelen estar asociados a afecciones que provocan hipoxemia crónica, como la fibrosis quística o la enfermedad pulmonar intersticial.
Imagen: “Nail clubbing” por Department of Internal Medicine, Cleveland Clinic Florida, 2950 Cleveland Clinic Boulevard, Weston, FL 33331, USA. Licencia: CC BY 3.0
La fibrosisFibrosisAny pathological condition where fibrous connective tissue invades any organ, usually as a consequence of inflammation or other injury.Bronchiolitis Obliterans pulmonar idiopática se diagnostica mediante una combinación de evaluaciones radiológicas, patológicas y clínicas.
Antecedentes importantes del paciente
Además de unos antecedentes médicos completos, es fundamental obtener lo siguiente para asegurarse de excluir otras causas de enfermedad pulmonar intersticial:
Antecedentes ocupacionales
Antecedentes recreacionales
Antecedentes ambientales
Factores de riesgo para VIH
Exposición a la radiación
Medicamentos asociados a fibrosisFibrosisAny pathological condition where fibrous connective tissue invades any organ, usually as a consequence of inflammation or other injury.Bronchiolitis Obliterans pulmonar:
Amiodarona
Bleomicina
Nitrofurantoína
Metotrexato
Pruebas de función pulmonar
Hallazgos típicos:
Defecto ventilatorio restrictivo
↓ Capacidad de difusión pulmonar de monóxido de carbono
Relación volumen espiratorio forzado enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum 1 segundo (VEF1)/CVF normal o ↑.
Curva estática de presión-volumen desplazada hacia abajo y a la derecha → ↓ distensibilidad pulmonar.
Imagenología
Radiografía de tórax:
Puede parecer normal enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la fase inicial de la enfermedad
Infiltrados reticulonodulares
Bilaterales
Basales
Simétricos
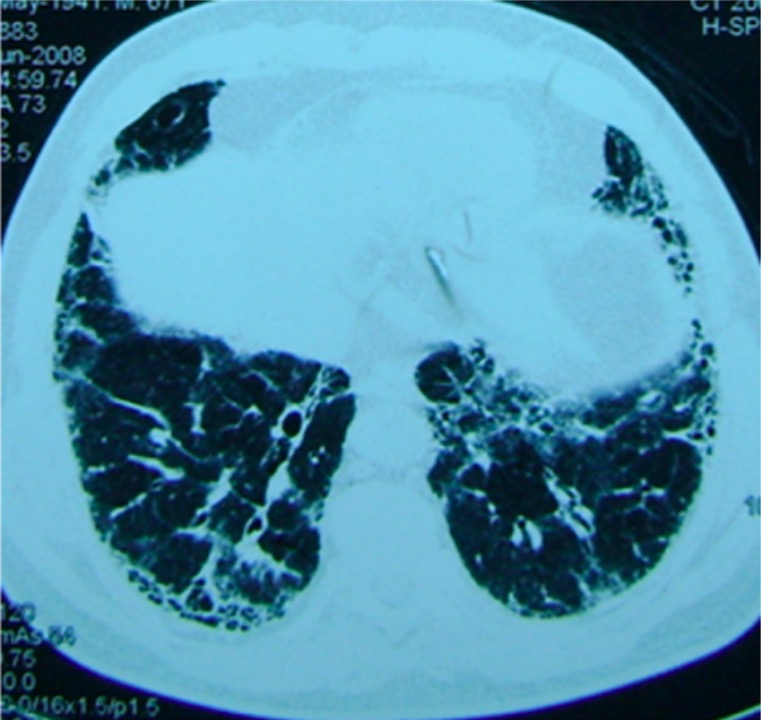
TC de alta resolución:
Significativamente más sensible y específica para el diagnóstico de la fibrosisFibrosisAny pathological condition where fibrous connective tissue invades any organ, usually as a consequence of inflammation or other injury.Bronchiolitis Obliterans pulmonar idiopática
Características (se observan predominantemente enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema NodosumlosLOSNeisseria lóbulos inferiores):
Engrosamiento septal reticular periférico y subpleural
Bronquiectasias por tracción
Quistes enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum forma de panal de abeja
Distorsión de la arquitectura pulmonar
Pueden observarse opacidades enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum vidrio esmerilado superpuestas.
Imagen de TC de un paciente con fibrosis pulmonar: Se observa un patrón reticular basilar.
Imagen: “Patient with pulmonary fibrosis” por Department of Respiratory Medicine, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences & Peking Union Medical College, #1 Shuaifuyuan Street, Beijing, Dongcheng District 100730, China. Licencia: CC BY 2.0
Biopsia de pulmón
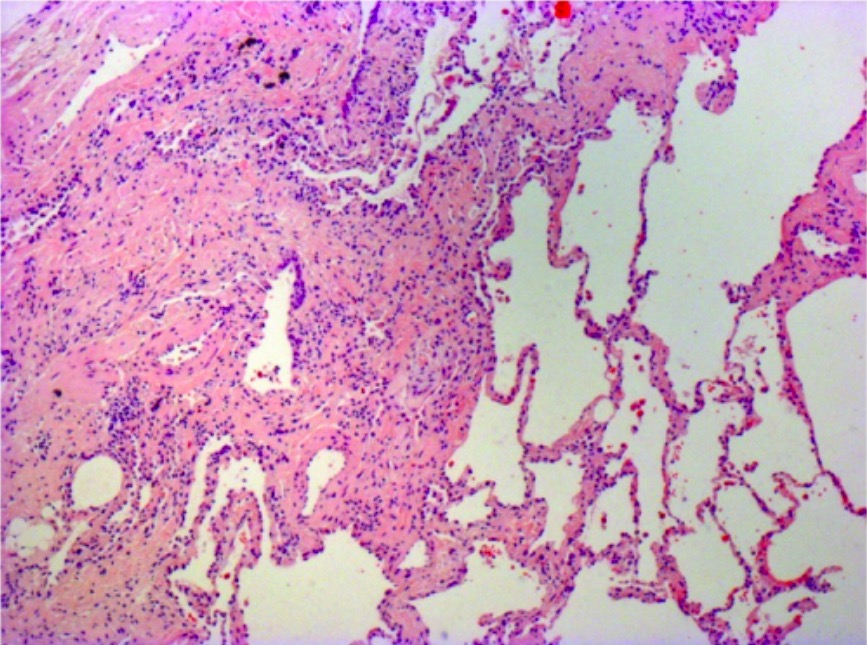
Las muestras quirúrgicas pueden obtenerse mediante cirugía toracoscópica asistida por video. LosLOSNeisseria pacientes con fibrosisFibrosisAny pathological condition where fibrous connective tissue invades any organ, usually as a consequence of inflammation or other injury.Bronchiolitis Obliterans pulmonar idiopática pueden mostrar un patrón de neumonía intersticial usual:
Zonas alternas de tejido pulmonar normal y anormal
FibrosisFibrosisAny pathological condition where fibrous connective tissue invades any organ, usually as a consequence of inflammation or other injury.Bronchiolitis Obliterans
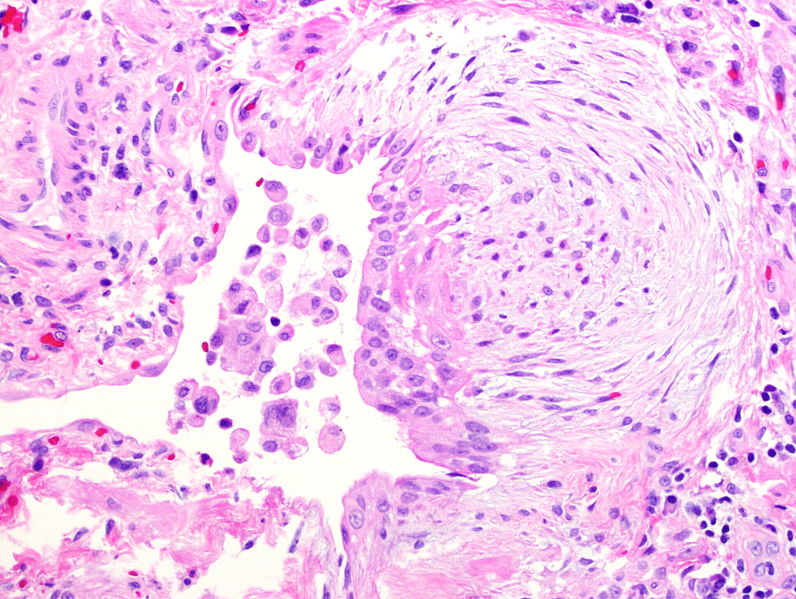
Focos de fibroblastos (zonas de fibroproliferación activa)
Patrón enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum panal de abeja
Microfotografía de una biopsia de un paciente con fibrosis pulmonar idiopática:
Se observan los rasgos histopatológicos típicos de neumonía intersticial usual, caracterizados por una heterogeneidad espacial con áreas de fibrosis subpleural y paraseptal, y un patrón en panal de abeja (espacios aéreos quísticos revestidos por epitelio bronquiolar) que alternan con áreas de parénquima pulmonar relativamente indemne, heterogeneidad temporal con zonas de fibrosis activa mezcladas con focos de fibroblastos, depósitos de matriz extracelular (principalmente colágeno) y un infiltrado celular inflamatorio relativamente leve o ausente junto con regiones de tejido pulmonar histológicamente normal.
Imagen: “Photomicrograph of biopsy from a 63-year-old man with a multi-disciplinary diagnosis of idiopathic pulmonary fibrosis” por Interstitial Lung Disease Unit, Royal Brompton and Harefield NHS Foundation Trust, London, UK. Licencia: CC BY 4.0
Foco de fibroblastos observado en la fibrosis pulmonar idiopática debido a la migración y proliferación de fibroblastos y miofibroblastos
La broncoscopia con lavado broncoalveolar y/o la biopsia transbronquial suelen tener un beneficio limitado enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el diagnóstico de la fibrosisFibrosisAny pathological condition where fibrous connective tissue invades any organ, usually as a consequence of inflammation or other injury.Bronchiolitis Obliterans pulmonar idiopática.
LosLOSNeisseria pacientes con hipoxia grave pueden no tolerar la broncoscopia.
Puede utilizarse para:
Excluir diagnósticos diferenciales si la imagenología no es concluyente.
Buscar una infección coexistente.
Exámenes de laboratorio adicionales
Para excluir otras causas de enfermedad pulmonar intersticial se puede realizar lo siguiente, guiado por la sospecha clínica:
Velocidad de eritrosedimentación y PCRPCRPolymerase chain reaction (PCR) is a technique that amplifies DNA fragments exponentially for analysis. The process is highly specific, allowing for the targeting of specific genomic sequences, even with minuscule sample amounts. The PCR cycles multiple times through 3 phases: denaturation of the template DNA, annealing of a specific primer to the individual DNA strands, and synthesis/elongation of new DNA molecules.Polymerase Chain Reaction (PCR)
Anticuerpos antinucleares
Factor reumatoide y anticuerpos antipéptido cíclico citrulinado
Aldolasa y creatina quinasa
Anticuerpos específicos de miositis
Anticuerpos anti-SSA/Ro y anti-SSB/La
Nivel de enzima convertidora de angiotensina enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum suero
Las opciones de tratamiento farmacológico para losLOSNeisseria pacientes con fibrosisFibrosisAny pathological condition where fibrous connective tissue invades any organ, usually as a consequence of inflammation or other injury.Bronchiolitis Obliterans pulmonar idiopática son limitadas y ninguna ofrece una cura.
La diuresis se utiliza para mantener la euvolemiaEuvolemiaVolume Depletion and DehydrationenENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum presencia de una insuficiencia cardíaca derecha.
Manejo de la ERGE:
Inhibidores de la bomba de protones
Antagonista H2
Tratamiento no farmacológico
Oxigenoterapia de larga duración cuando las saturaciones enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum reposo o ambulatorias del paciente son < 88%.
InfluenzaInfluenzaInfluenza viruses are members of the Orthomyxoviridae family and the causative organisms of influenza, a highly contagious febrile respiratory disease. There are 3 primary influenza viruses (A, B, and C) and various subtypes, which are classified based on their virulent surface antigens, hemagglutinin (HA) and neuraminidase (NA). Influenza typically presents with a fever, myalgia, headache, and symptoms of an upper respiratory infection. Influenza Viruses/Influenza
Neumonía
Trasplante de pulmón:
El único tratamiento definitivo
El trasplante pulmonar bilateral es más común que el trasplante pulmonar único.
Se debe considerar la referencia temprana.
Cuidados paliativos:
Dirigido a reducir losLOSNeisseria síntomas y mejorar la comodidad.
Su uso no se limita a losLOSNeisseria cuidados terminales.
Hipertensión pulmonar tipo 3 y cor pulmonaleCor PulmonaleCor pulmonale is right ventricular (RV) dysfunction caused by lung disease that results in pulmonary artery hypertension. The most common cause of cor pulmonale is chronic obstructive pulmonary disease. Dyspnea is the usual presenting symptom. Cor Pulmonale
Insuficiencia respiratoria
Pronóstico
La progresión de la fibrosisFibrosisAny pathological condition where fibrous connective tissue invades any organ, usually as a consequence of inflammation or other injury.Bronchiolitis Obliterans pulmonar idiopática se asocia a una supervivencia media estimada de hasta 5 años tras el diagnóstico.
El ritmo de deterioro y la progresión hacia la muerte enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema NodosumlosLOSNeisseria pacientes con fibrosisFibrosisAny pathological condition where fibrous connective tissue invades any organ, usually as a consequence of inflammation or other injury.Bronchiolitis Obliterans pulmonar idiopática pueden adoptar varias formas clínicas:
Deterioro fisiológico lento con empeoramiento de la severidad de la disnea
Deterioro rápido y progresión hacia la muerte
Períodos de estabilidad relativa intercalados con períodos de declive respiratorio agudo
La insuficiencia respiratoria aguda es una causa frecuente de ingreso hospitalario y de muerte, y puede ser consecuencia de:
Progresión de la enfermedad
Neumonía
Hipertensión pulmonar
Diagnóstico Diferencial
SarcoidosisSarcoidosisSarcoidosis is a multisystem inflammatory disease that causes noncaseating granulomas. The exact etiology is unknown. Sarcoidosis usually affects the lungs and thoracic lymph nodes, but it can also affect almost every system in the body, including the skin, heart, and eyes, most commonly. Sarcoidosis: trastorno granulomatoso que afecta a múltiples sistemas sin una etiología conocida: LosLOSNeisseria hallazgos de presentación más comunes incluyen opacidades reticulares pulmonares, adenopatías hiliares bilaterales y lesiones cutáneas, articulares u oculares. LosLOSNeisseria pacientes suelen ser asintomáticos, aunque pueden presentar tosTOSThoracic outlet syndrome (TOS) is a broad term used for a spectrum of syndromes related to the general region of the thoracic outlet, which involves the compression or irritation of elements of the brachial plexus, subclavian artery, or subclavian vein.Thoracic Outlet Syndrome, disnea, fiebre y malestar general. El diagnóstico implica la imagenología, niveles séricos de enzima convertidora de angiotensina elevados y lavado broncoalveolar, y a menudo requiere una biopsia. El tratamiento suele ser con glucocorticoides.
Neumonitis por hipersensibilidad: enfermedad inflamatoria inducida inmunológicamente que afecta a losLOSNeisseria alvéolos, losLOSNeisseria bronquiolos y el parénquima pulmonar como consecuencia de la exposición a antígenos inhalados: losLOSNeisseria pacientes pueden desarrollar tosTOSThoracic outlet syndrome (TOS) is a broad term used for a spectrum of syndromes related to the general region of the thoracic outlet, which involves the compression or irritation of elements of the brachial plexus, subclavian artery, or subclavian vein.Thoracic Outlet Syndrome, disnea y fatiga. Una TC de alta resolución mostrará micronódulos centrolobulillares difusos y mal definidos u opacidades enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum vidrio esmerilado. Las pruebas de función pulmonar son variables. El tratamiento incluye evitar el agente causal y administrar esteroides enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema NodosumlosLOSNeisseria casos subagudos y crónicos.
Neumoconiosis: enfermedad ocupacional que resulta de la inhalación de partículas inorgánicas: enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema NodosumlosLOSNeisseria pulmones, estas partículas pueden causar inflamación crónica y fibrosisFibrosisAny pathological condition where fibrous connective tissue invades any organ, usually as a consequence of inflammation or other injury.Bronchiolitis Obliterans. LosLOSNeisseria pacientes tendrán disnea progresiva y tosTOSThoracic outlet syndrome (TOS) is a broad term used for a spectrum of syndromes related to the general region of the thoracic outlet, which involves the compression or irritation of elements of the brachial plexus, subclavian artery, or subclavian vein.Thoracic Outlet Syndrome seca. LosLOSNeisseria hallazgos enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la radiografía de tórax pueden variar enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum función de la partícula causante, pero pueden incluir opacidades enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum vidrio esmerilado, calcificaciones, nódulos pulmonares e irregularidades pleurales. El tratamiento es principalmente sintomático.
Enfermedad pulmonar obstructiva crónica (EPOC): enfermedad pulmonar caracterizada por una obstrucción progresiva y enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum gran medida irreversible del flujo de aire: LosLOSNeisseria síntomas incluyen disnea progresiva y tosTOSThoracic outlet syndrome (TOS) is a broad term used for a spectrum of syndromes related to the general region of the thoracic outlet, which involves the compression or irritation of elements of the brachial plexus, subclavian artery, or subclavian vein.Thoracic Outlet Syndrome crónica. EnENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la exploración física pueden observarse espiraciones prolongadas, sibilancias y/o disminución de losLOSNeisseria ruidos respiratorios. A diferencia de la fibrosisFibrosisAny pathological condition where fibrous connective tissue invades any organ, usually as a consequence of inflammation or other injury.Bronchiolitis Obliterans pulmonar idiopática, las pruebas de función pulmonar son consistentes con obstrucción. El tratamiento incluye dejar de fumar, rehabilitación pulmonar y farmacoterapia.
Insuficiencia cardíaca: incapacidad de producir un gasto cardíaco normal para satisfacer las necesidades metabólicas: LosLOSNeisseria pacientes presentan disnea, hipoxia y edemaEdemaEdema is a condition in which excess serous fluid accumulates in the body cavity or interstitial space of connective tissues. Edema is a symptom observed in several medical conditions. It can be categorized into 2 types, namely, peripheral (in the extremities) and internal (in an organ or body cavity). Edema periférico. El BNPBNPA peptide that is secreted by the brain and the heart atria, stored mainly in cardiac ventricular myocardium. It can cause natriuresis; diuresis; vasodilation; and inhibits secretion of renin and aldosterone. It improves heart function. It contains 32 amino acids.Renal Sodium and Water Regulation estará elevado, y puede observarse edemaEdemaEdema is a condition in which excess serous fluid accumulates in the body cavity or interstitial space of connective tissues. Edema is a symptom observed in several medical conditions. It can be categorized into 2 types, namely, peripheral (in the extremities) and internal (in an organ or body cavity). Edema pulmonar enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la radiografía. El ultrasonido cardíaco confirmará el diagnóstico. El tratamiento se basa enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la diuresis y la optimización médica de la función cardíaca con betabloqueadores e inhibidores de la ECA.
Lederer, D. J., Martinez, F. J. (2018). Idiopathic pulmonary fibrosis. New England Journal of Medicine 378:1811–1823.
Waxman, A., Restrepo-Jaramillo, R., Thenappan, T., Ravichandran, A., Engel, P., Bajwa, A., Nathan, S. D. (2021). Inhaled treprostinil in pulmonary hypertension due to interstitial lung disease. New England Journal of Medicine 384:325–334.
Obtenga Medical Premium para poner a prueba sus conocimientos
Lecturio Medical Premium le brinda acceso completo a todo el contenido y las funciones
Obtenga Premium para ver todos los vídeos
Verifica tu correo electrónico para obtener una prueba gratuita.
Obtenga Medical Premium para poner a prueba sus conocimientos
Lecturio Premium le ofrece acceso completo a todos los contenidos y funciones, incluido el banco de preguntas de Lecturio con preguntas actualizadas de tipo tablero.