La anemia Anemia Anemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types drepanocítica o de células falciformes es un grupo de trastornos genéticos en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum los LOS Neisseria que una molécula de hemoglobina ( Hb Hb The oxygen-carrying proteins of erythrocytes. They are found in all vertebrates and some invertebrates. The number of globin subunits in the hemoglobin quaternary structure differs between species. Structures range from monomeric to a variety of multimeric arrangements. Gas Exchange S) anormal transforma los LOS Neisseria eritrocitos en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum células falciformes, lo que provoca anemia Anemia Anemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types crónica, episodios vasooclusivos, dolor Dolor Inflammation y daño de órganos. El rasgo de células falciformes, que es la afección heterocigota, es el único del grupo que generalmente es benigno y rara vez se asocia con complicaciones graves similares a las de la anemia Anemia Anemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types de células falciformes. Desencadenantes como el estrés y la hipoxia pueden inducir o empeorar la formación de eritrocitos falciformes. Los LOS Neisseria individuos con anemia Anemia Anemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types de células falciformes son susceptibles a infecciones, infartos de varios órganos y aplasia Aplasia Cranial Nerve Palsies de la médula ósea; la afectación pulmonar en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el síndrome torácico agudo puede ser rápidamente letal. Las células falciformes generalmente se pueden ver en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el frotis de sangre periférica, pero se necesita la electroforesis de hemoglobina para el diagnóstico. El tratamiento de los LOS Neisseria episodios dolorosos consiste en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum líquidos y analgésicos por vía intravenosa y, en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum episodios graves, puede ser necesaria la exanguinotransfusión. La supervivencia mejora con la vacunación contra las infecciones bacterianas, los LOS Neisseria antibióticos profilácticos y el tratamiento agresivo de las infecciones.
Last updated: Dec 15, 2025
La anemia Anemia Anemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types drepanocítica o de células falciformes es un grupo de trastornos genéticos que causan una molécula de hemoglobina ( Hb Hb The oxygen-carrying proteins of erythrocytes. They are found in all vertebrates and some invertebrates. The number of globin subunits in the hemoglobin quaternary structure differs between species. Structures range from monomeric to a variety of multimeric arrangements. Gas Exchange S) anormal que transforma los LOS Neisseria eritrocitos en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum células falciformes, lo que resulta en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum anemia Anemia Anemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types crónica, episodios vasooclusivos, dolor Dolor Inflammation y daño de órganos.
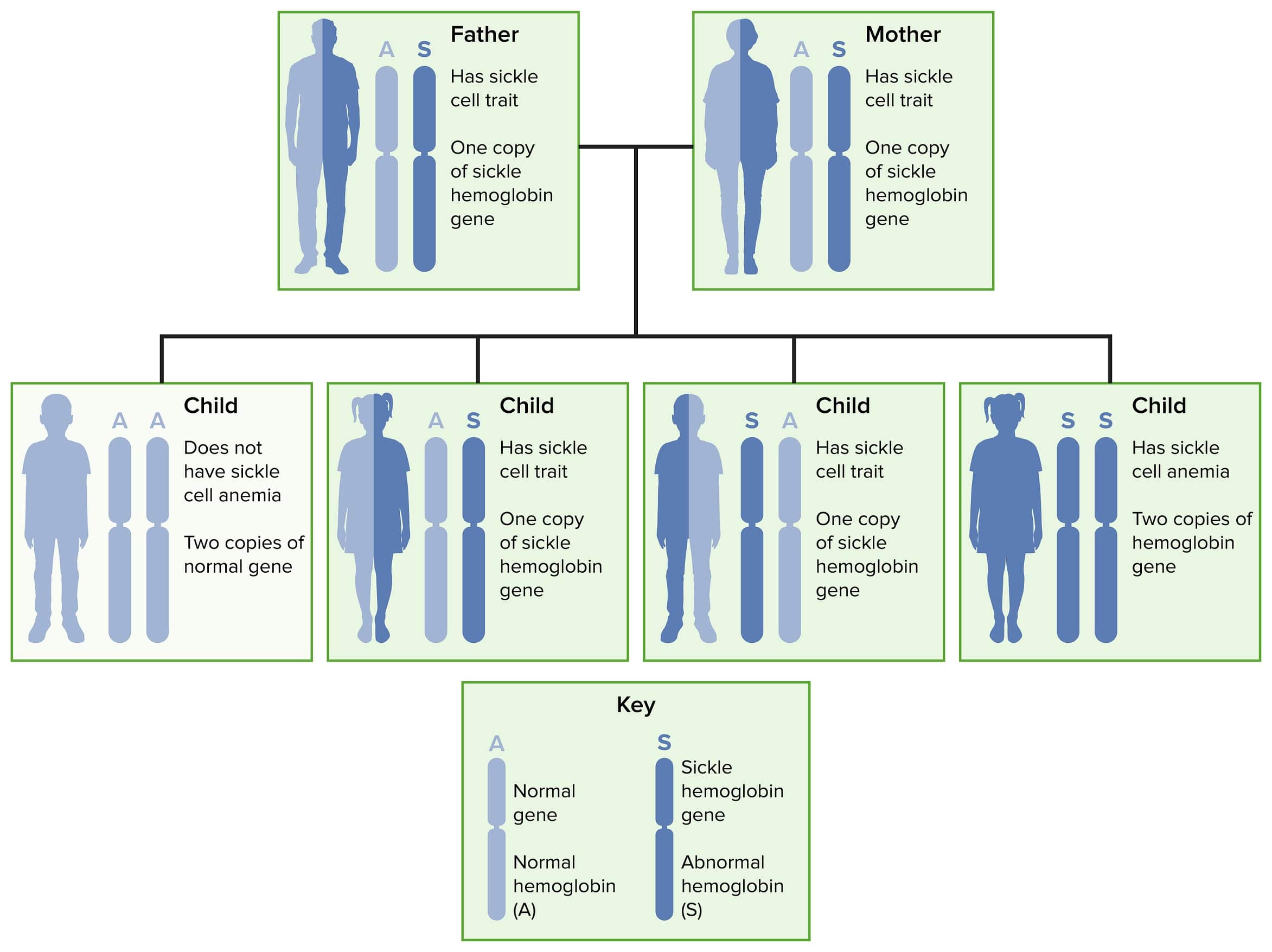
Herencia autosómica recesiva de la enfermedad de células falciformes y su rasgo
Imagen por Lecturio.La molécula normal de hemoglobina adulta (HbA1) consta de 2 pares de cadenas llamadas alfa y beta.
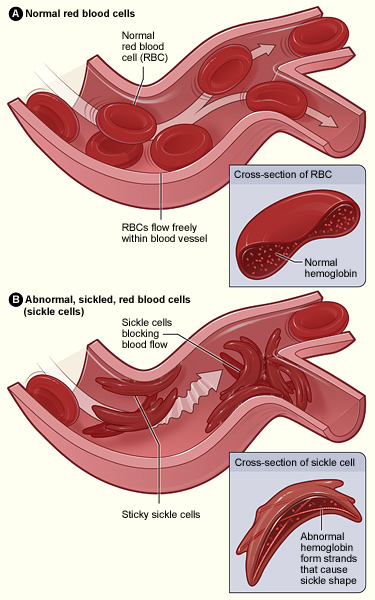
La hemoglobina anormal da lugar a la formación de hongos en los glóbulos rojos y a la adhesión de las células falciformes al endotelio, que es activado por los glóbulos rojos adheridos. La oclusión de los vasos pequeños se produce por un agregado de glóbulos rojos falciformes, con plaquetas y glóbulos blancos (no se muestra en la figura).
Imagen: “Sickle cell 01” por The National Heart, Lung, and Blood Institute (NHLBI). Licencia: Dominio PúblicoLa mayoría de los LOS Neisseria síntomas resultan de la anemia Anemia Anemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types y los LOS Neisseria eventos vasooclusivos que se observan en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum individuos con anemia Anemia Anemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types drepanocítica o complicaciones que incluyen infección.
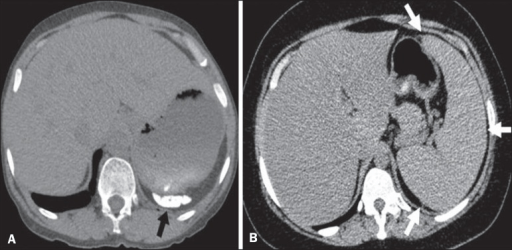
Diferentes patrones de afectación esplénica en la enfermedad de células falciformes:
A: atrofia y calcificación del bazo (flecha)
B: esplenomegalia (flechas)
La enfermedad de células falciformes generalmente se diagnostica prenatalmente o al AL Amyloidosis nacer mediante un tamizaje neonatal obligatorio. Los LOS Neisseria métodos varían de un estado a otro.
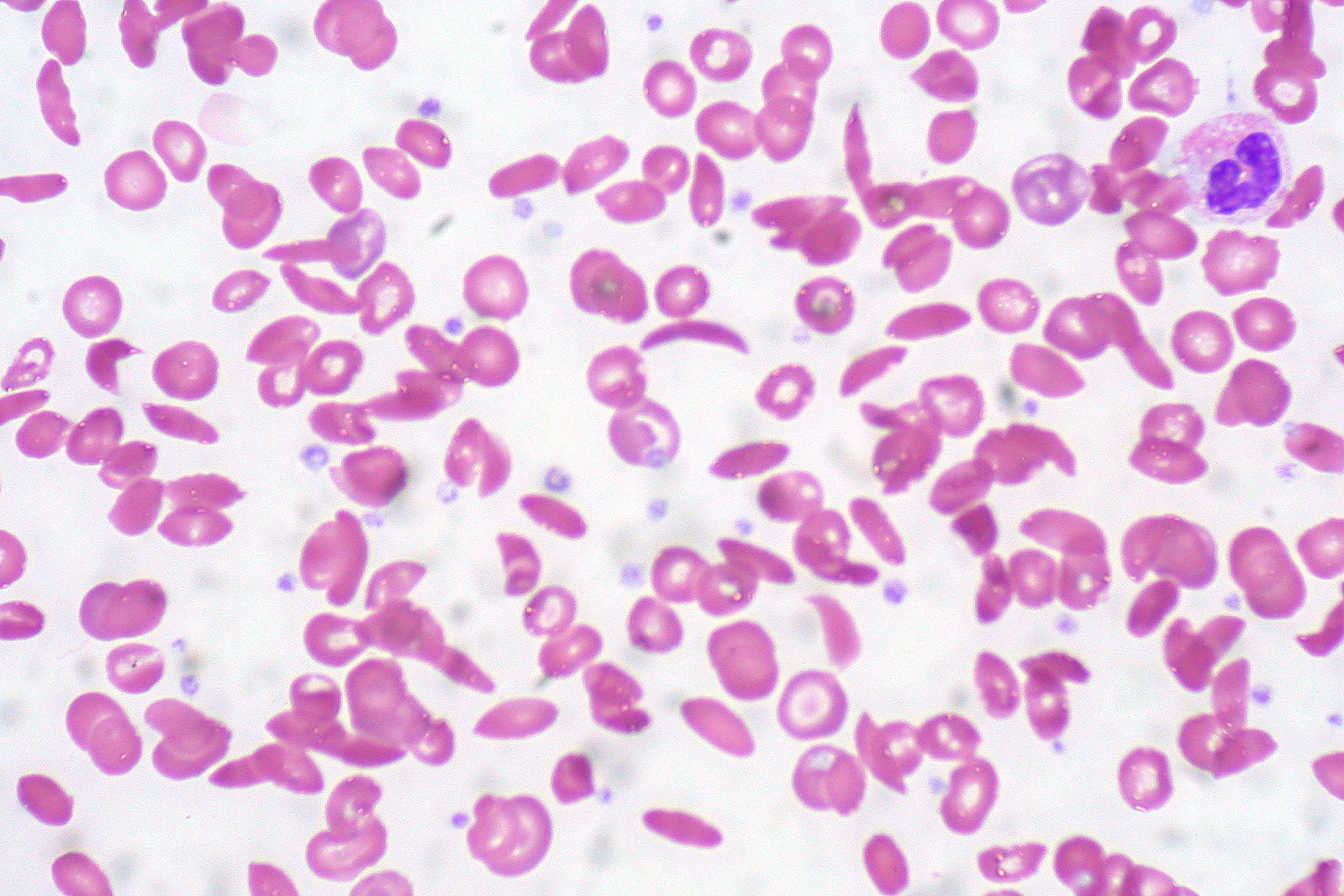
Frotis de sangre periférica que muestra una mezcla de eritrocitos, algunos con morfología redonda normal y otros con forma de hoz (alargamiento y flexión)
Imagen: “Sickle Cell Anemia” por Ed Uthman. Licencia: CC BY 2.0El tratamiento de los LOS Neisseria episodios dolorosos incluye analgésicos y medidas generales de soporte. Las transfusiones pueden ser necesarias en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum ocasiones si el paciente tiene anemia Anemia Anemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types sintomática, incluyendo la complicación del síndrome torácico agudo
| Presentación clínica | Tratamiento |
|---|---|
| Episodios de dolor Dolor Inflammation agudo/eventos vasooclusivos |
|
| Secuestro esplénico agudo |
|
| Infecciones | Prevención:
|
| Priapismo |
|
| Tamizaje profiláctico |
|
| Refractario |
|