Lysosomal storage diseases are a group of metabolic disorders caused by genetic mutations Genetic Mutations Carcinogenesis in the enzymes Enzymes Enzymes are complex protein biocatalysts that accelerate chemical reactions without being consumed by them. Due to the body's constant metabolic needs, the absence of enzymes would make life unsustainable, as reactions would occur too slowly without these molecules. Basics of Enzymes responsible for normal lysosomal function. The dysfunction of enzymatic processes causes an accumulation of undigested metabolites, resulting in cellular death. The main groups of lysosomal storage diseases include sphingolipidoses, oligosaccharidoses, and mucolipidoses. Subgroups within the main diseases have different underlying mechanisms and clinical manifestations.
Last updated: Apr 24, 2025
Lysosomal storage diseases are rare metabolic conditions caused by genetic mutations Genetic Mutations Carcinogenesis of lysosomal enzymes Enzymes Enzymes are complex protein biocatalysts that accelerate chemical reactions without being consumed by them. Due to the body’s constant metabolic needs, the absence of enzymes would make life unsustainable, as reactions would occur too slowly without these molecules. Basics of Enzymes, which lead to dysfunctional metabolism and the accumulation of glycosaminoglycans, glycoproteins Glycoproteins Conjugated protein-carbohydrate compounds including mucins, mucoid, and amyloid glycoproteins. Basics of Carbohydrates, or glycolipids Glycolipids Lipid attached to carbohydrate, outward-facing. The Cell: Cell Membrane.
Disorders are due to a deficiency in a specific lysosomal hydrolase or the enzymes Enzymes Enzymes are complex protein biocatalysts that accelerate chemical reactions without being consumed by them. Due to the body’s constant metabolic needs, the absence of enzymes would make life unsustainable, as reactions would occur too slowly without these molecules. Basics of Enzymes required for lysosomal function:
The disorders are considered as groups of individually rare inherited disorders of intracellular metabolism. Of the 40 classified disorders, 15 account for the majority of cases. Categorization Categorization Types of Variables is by accumulated metabolite intermediates:
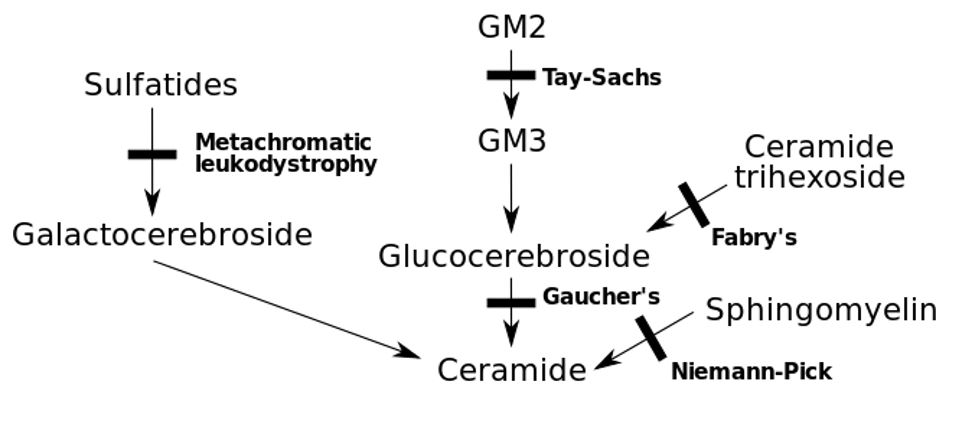
Inborn errors of metabolism with the associated genetic deficit
Image: “Inborn errors of metabolism” by Huckfinne. License: Public Domain| Group | Subgroup | Description |
|---|---|---|
| Sphingolipidoses | GM2-gangliosidosis |
|
| GM1-gangliosidosis | Caused by a mutation Mutation Genetic mutations are errors in DNA that can cause protein misfolding and dysfunction. There are various types of mutations, including chromosomal, point, frameshift, and expansion mutations. Types of Mutations in the GLB1 gene Gene A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. Basic Terms of Genetics encoding for β-galactosidase-1 | |
| Gaucher disease Gaucher disease Gaucher Disease (GD) is an autosomal recessive lysosomal storage disorder caused by a deficiency of glucocerebrosidase enzyme activity, resulting in accumulation of glucocerebroside in cells and certain organs. The disease is categorized into 3 types with variable clinical presentation. Gaucher Disease |
|
|
| Fabry disease Fabry disease Fabry disease (FD), also known as Anderson-Fabry disease, is an X-linked recessive lysosomal storage disorder and the 2nd most common of the lysosomal storage disorders. Fabry disease is caused by a deficiency in the alpha-galactosidase enzyme (alpha-Gal A), resulting in the accumulation of the glycosphingolipid globotriaosylceramide (Gb3) in lysosomes. Fabry Disease |
|
|
| Metachromatic leukodystrophy Metachromatic leukodystrophy Metachromatic leukodystrophy (MLD) is an inherited lysosomal storage disorder that affects myelin in the brain and spinal cord. Genetic mutations result in the creation of a dysfunctional arylsulfatase A (ARSA) enzyme, which is unable to break down cerebroside sulfate. The accumulation of this metabolite results in permanent damage to oligodendroglial and Schwann cells (myelin). Metachromatic Leukodystrophy | Deficiency in arylsulfatase A activity | |
| Krabbe disease Krabbe disease Krabbe disease, also known as globoid cell leukodystrophy or galactosylceramide lipidosis, is a rare autosomal recessive lysosomal storage disorder caused by a deficiency of the enzyme galactocerebrosidase. Accumulation of galactocerebroside results in destruction of myelin-producing cells throughout the peripheral and central nervous systems, leading to demyelination and clinical symptoms. Krabbe Disease |
|
|
| Disseminated lipogranulomatosis (Farber disease) |
|
|
| Niemann–Pick disease |
|
|
| Oligosaccharidoses | Sialidosis |
|
| Galactosialidosis |
|
|
| Fucosidosis |
|
|
| Mannosidosis | 2 types:
|
|
| Mucolipidoses | I-cell disease I-cell disease Inclusion-cell disease (I-cell disease, mucolipidosis II, or ML II) is caused by a defect in uridine diphosphate (UDP)-N-acetylglucosamine-1-phosphotransferase, an enzyme that transfers phosphate to mannose residues on specific proteins. This protein is essential, since it is responsible for the breakdown of oligosaccharides, lipids, and glycosaminoglycans. I-cell Disease |
|
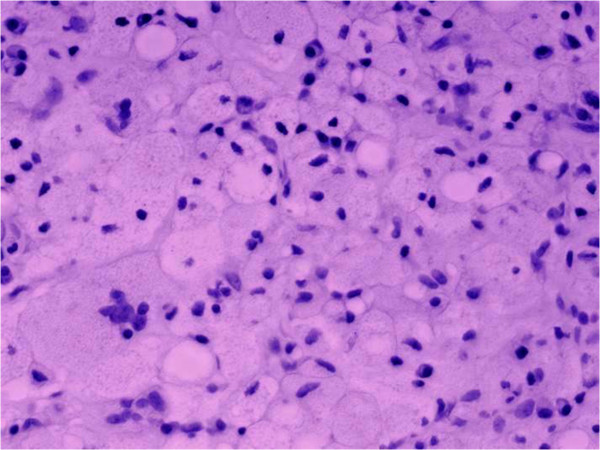
Gaucher disease leads to bone necrosis:
Connective tissue is infiltrated with numerous Gaucher cells (vacuolated, lipid-laden reticuloendothelial cells with enlarged granular cytoplasm and round, displaced nuclei).
The presentation is variable Variable Variables represent information about something that can change. The design of the measurement scales, or of the methods for obtaining information, will determine the data gathered and the characteristics of that data. As a result, a variable can be qualitative or quantitative, and may be further classified into subgroups. Types of Variables depending upon the etiology of the lysosomal storage disorder and may occur soon after birth or late into adulthood:
| Group | Subgroup | Signs and symptoms |
|---|---|---|
| Sphingolipidoses | GM2-gangliosidosis |
|
| GM1-gangliosidosis |
|
|
| Gaucher disease Gaucher disease Gaucher Disease (GD) is an autosomal recessive lysosomal storage disorder caused by a deficiency of glucocerebrosidase enzyme activity, resulting in accumulation of glucocerebroside in cells and certain organs. The disease is categorized into 3 types with variable clinical presentation. Gaucher Disease |
|
|
| Fabry disease Fabry disease Fabry disease (FD), also known as Anderson-Fabry disease, is an X-linked recessive lysosomal storage disorder and the 2nd most common of the lysosomal storage disorders. Fabry disease is caused by a deficiency in the alpha-galactosidase enzyme (alpha-Gal A), resulting in the accumulation of the glycosphingolipid globotriaosylceramide (Gb3) in lysosomes. Fabry Disease |
|
|
| Metachromatic leukodystrophy Metachromatic leukodystrophy Metachromatic leukodystrophy (MLD) is an inherited lysosomal storage disorder that affects myelin in the brain and spinal cord. Genetic mutations result in the creation of a dysfunctional arylsulfatase A (ARSA) enzyme, which is unable to break down cerebroside sulfate. The accumulation of this metabolite results in permanent damage to oligodendroglial and Schwann cells (myelin). Metachromatic Leukodystrophy |
|
|
| Krabbe disease Krabbe disease Krabbe disease, also known as globoid cell leukodystrophy or galactosylceramide lipidosis, is a rare autosomal recessive lysosomal storage disorder caused by a deficiency of the enzyme galactocerebrosidase. Accumulation of galactocerebroside results in destruction of myelin-producing cells throughout the peripheral and central nervous systems, leading to demyelination and clinical symptoms. Krabbe Disease |
|
|
| Niemann-Pick disease Niemann-Pick disease Niemann-Pick disease (NPD) is a rare, inherited, lysosomal storage disorder. The disease is classified on the basis of the genetic mutation. Type A and type B result from mutations in the SMPD-1 gene, resulting in acid sphingomyelinase enzyme deficiency. Type C results from NPC1 or NPC2 gene mutations, which are needed for intracellular transport of lipids. Niemann-Pick Disease |
|
|
| Oligosaccharidoses | Sialidosis |
|
| Galactosialidosis |
|
|
| Fucosidosis |
|
|
| Mucolipidoses | I-cell disease I-cell disease Inclusion-cell disease (I-cell disease, mucolipidosis II, or ML II) is caused by a defect in uridine diphosphate (UDP)-N-acetylglucosamine-1-phosphotransferase, an enzyme that transfers phosphate to mannose residues on specific proteins. This protein is essential, since it is responsible for the breakdown of oligosaccharides, lipids, and glycosaminoglycans. I-cell Disease |
|
Work-up is based on the specific lysosomal storage disease, but some general principles are important to understand:
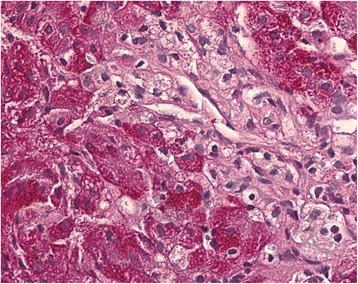
A Niemann-Pick cell from a liver sample showing swollen Kupffer cells with foamy cytoplasm, which is typical for Niemann-Pick disease type C
Image: “Histopathological liver biopsy findings” by Degtyareva AV, Mikhailova SV, Zakharova EY, Tumanova EL, Puchkova AA. License: CC BY 4.0Management depends upon the specific disorder. The general principles of management include:
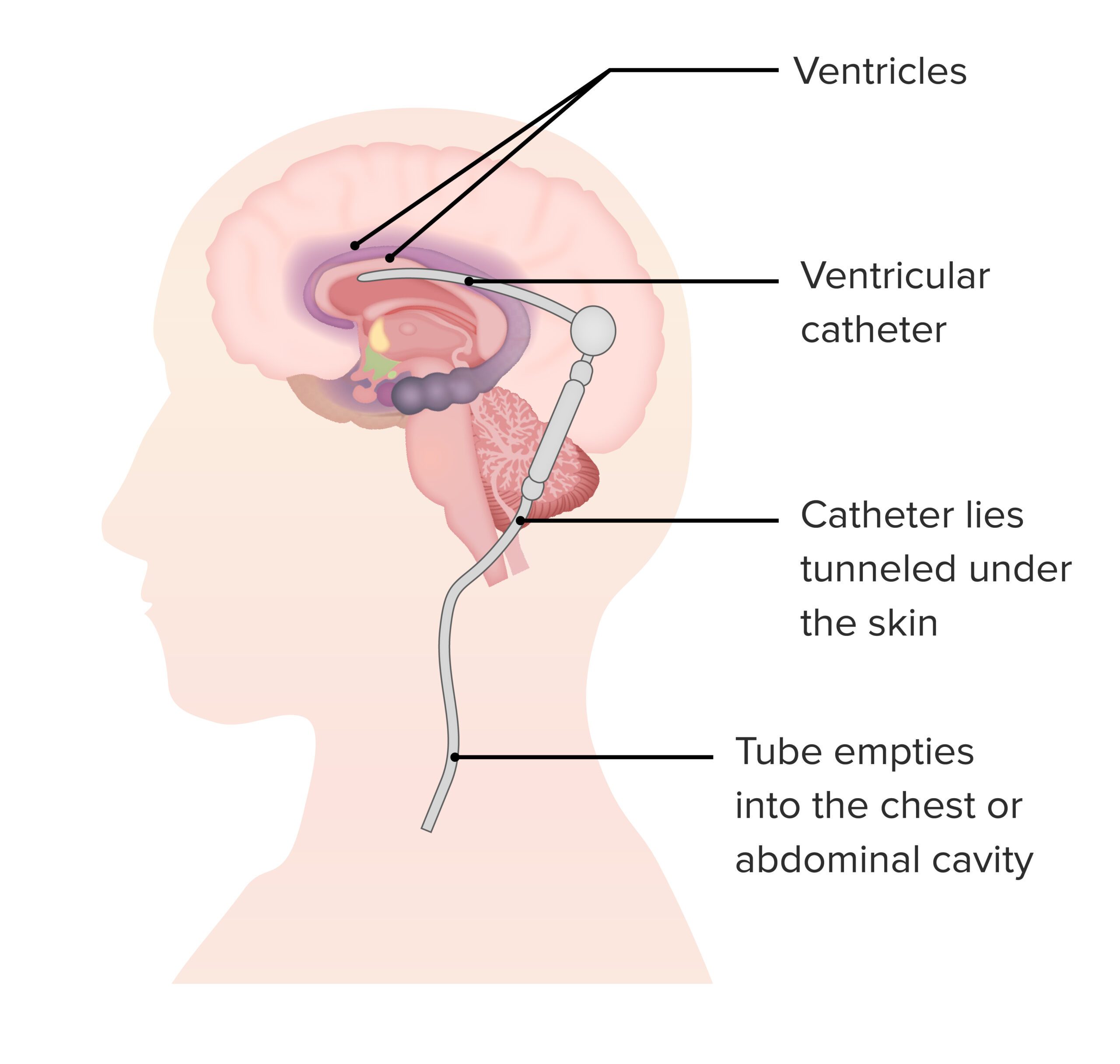
A shunt may be required for individuals with hydrocephalus due to a lysosomal storage disorder.
Image by Lecturio.While lysosomal storage disorders encompass a broad spectrum Broad Spectrum Macrolides and Ketolides of disease, various other conditions may overlap the disorders. Some conditions are subgroups within the umbrella of lysosomal storage disorders: