Enzymes are complex protein biocatalysts that accelerate chemical reactions without being consumed by them. Due to the body's constant metabolic needs, the absence of enzymes would make life unsustainable, as reactions would occur too slowly without these molecules. Enzymes have many functions, including macromolecule degradation and synthesis Synthesis Polymerase Chain Reaction (PCR) (catabolism and anabolism), signal transduction Transduction The transfer of bacterial DNA by phages from an infected bacterium to another bacterium. This also refers to the transfer of genes into eukaryotic cells by viruses. This naturally occurring process is routinely employed as a gene transfer technique. Bacteriology, energy generation ( adenosine Adenosine A nucleoside that is composed of adenine and d-ribose. Adenosine or adenosine derivatives play many important biological roles in addition to being components of DNA and RNA. Adenosine itself is a neurotransmitter. Class 5 Antiarrhythmic Drugs triphosphate [ATP]), ion pumps/ active transport Active transport The movement of materials across cell membranes and epithelial layers against an electrochemical gradient, requiring the expenditure of metabolic energy. The Cell: Cell Membrane, defense and clearance reactions (oxidization, reduction, hydrolysis Hydrolysis The process of cleaving a chemical compound by the addition of a molecule of water. Proteins and Peptides), cell regulation, movement ( myosin Myosin A diverse superfamily of proteins that function as translocating proteins. They share the common characteristics of being able to bind actins and hydrolyze mgATP. Myosins generally consist of heavy chains which are involved in locomotion, and light chains which are involved in regulation. Within the structure of myosin heavy chain are three domains: the head, the neck and the tail. The head region of the heavy chain contains the actin binding domain and mgATPase domain which provides energy for locomotion. The neck region is involved in binding the light-chains. The tail region provides the anchoring point that maintains the position of the heavy chain. The superfamily of myosins is organized into structural classes based upon the type and arrangement of the subunits they contain. Skeletal Muscle Contraction ATPase, transport of intracellular substances), and immune responses.
Last updated: Apr 23, 2025
An enzyme is a protein that presents active sites which perform reactions by decreasing the activation energy of that reaction.
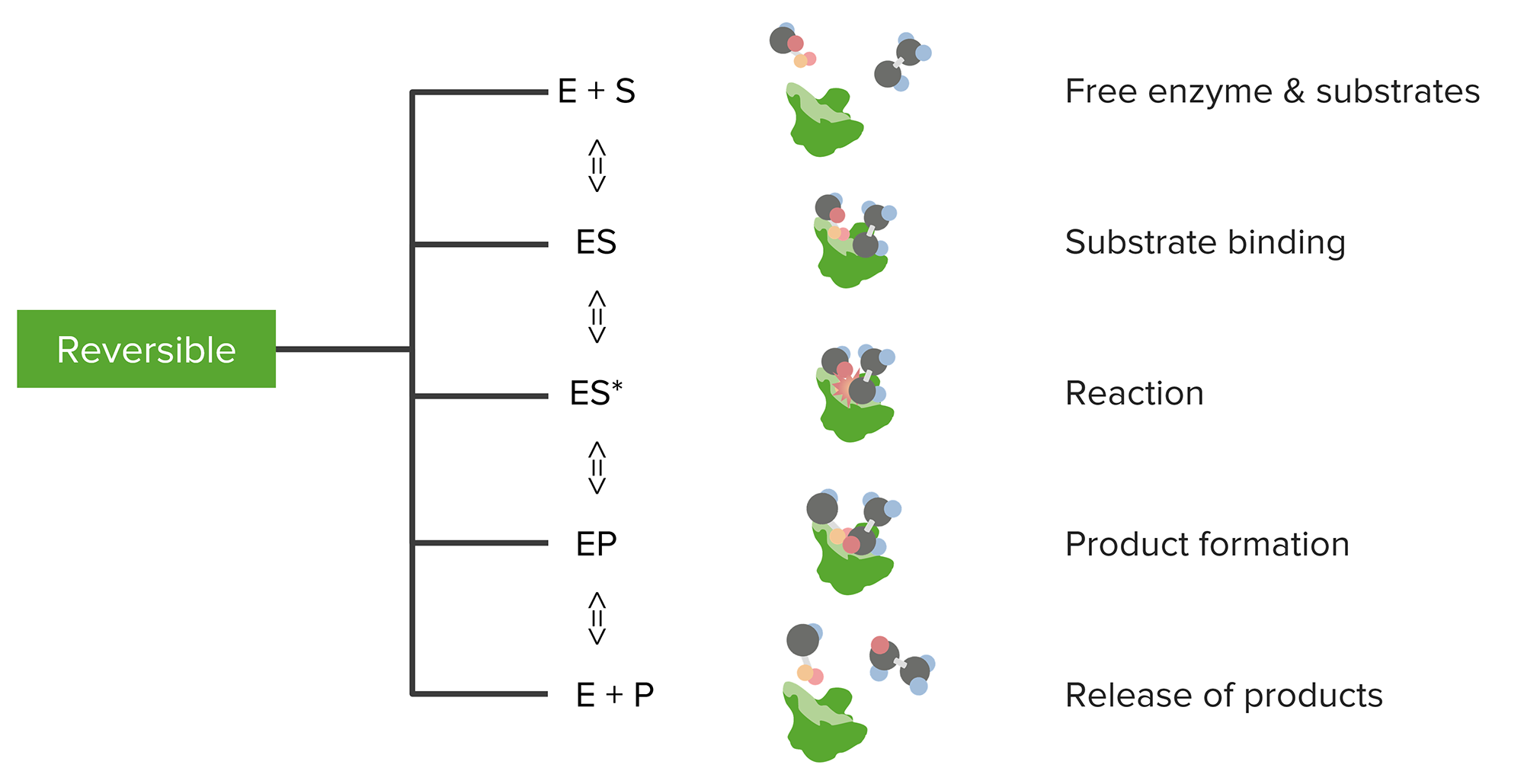
Relationship between substrates and enzymes, the reversible enzymatic reaction, and product formation and release
Image by Lecturio.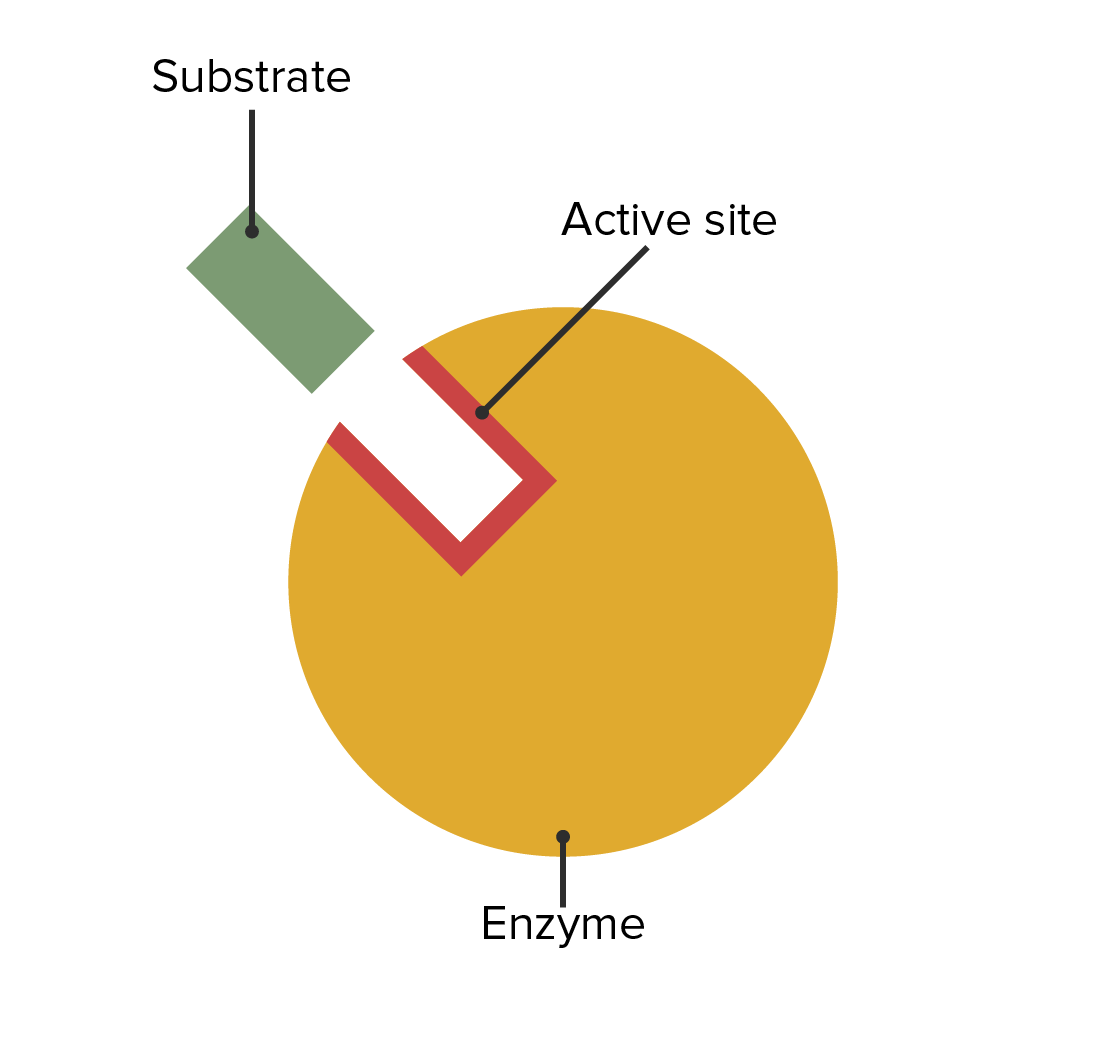
The location of the enzymatic active site and its relation with the substrate
Image by Lecturio.The first part of the name describes the substrate. The last part describes the enzyme function. The first part will describe the product if the following are true:
| Main group | Catalytic reaction | Important subclasses (examples) |
|---|---|---|
| Oxidoreductases | Transfer of reduction equivalents (1 mole Mole Nevi (singular nevus), also known as “moles,” are benign neoplasms of the skin. Nevus is a non-specific medical term because it encompasses both congenital and acquired lesions, hyper- and hypopigmented lesions, and raised or flat lesions. Nevus/Nevi of electrons); the electron donor is oxidized and increases its charge, the electron acceptor is reduced and decreases its charge | Dehydrogenases ( alcohol dehydrogenase Alcohol dehydrogenase A zinc-containing enzyme which oxidizes primary and secondary alcohols or hemiacetals in the presence of nad. In alcoholic fermentation, it catalyzes the final step of reducing an aldehyde to an alcohol in the presence of nadh and hydrogen. Ethanol Metabolism), oxidases (xanthine oxidase Oxidase Neisseria), reductases (glutathione reductase) |
| Transferases | Transfer of entire groups (e.g., amino groups) |
|
| Hydrolases | Molecular fission with water addition = hydrolytic fission |
|
| Lyases | Break bonds between 2 carbons, or a carbon atom and oxygen, or carbon and sulfur |
|
| Isomerases | Conversion of isomeric molecules into each other without changing the molecular formula | Cis-trans isomerases (peptidyl-prolyl cis-trans isomerase, phosphoglucoisomerase Phosphoglucoisomerase An aldose-ketose isomerase that catalyzes the reversible interconversion of glucose 6-phosphate and fructose 6-phosphate. In prokaryotic and eukaryotic organisms it plays an essential role in glycolytic and gluconeogenic pathways. In mammalian systems the enzyme is found in the cytoplasm and as a secreted protein. This secreted form of glucose-6-phosphate isomerase has been referred to as autocrine motility factor or neuroleukin, and acts as a cytokine which binds to the autocrine motility factor receptor. Deficiency of the enzyme in humans is an autosomal recessive trait, which results in congenital nonspherocytic hemolytic anemia. Gluconeogenesis) |
| Ligases | Also called synthetases, energy-dependent linkage of compounds (e.g., dependent on ATP) |
|
Enzymes can be modified in common ways to allow different organs to have the same activities or for substances outside of the substrate/enzyme/product sequence to influence enzymes.
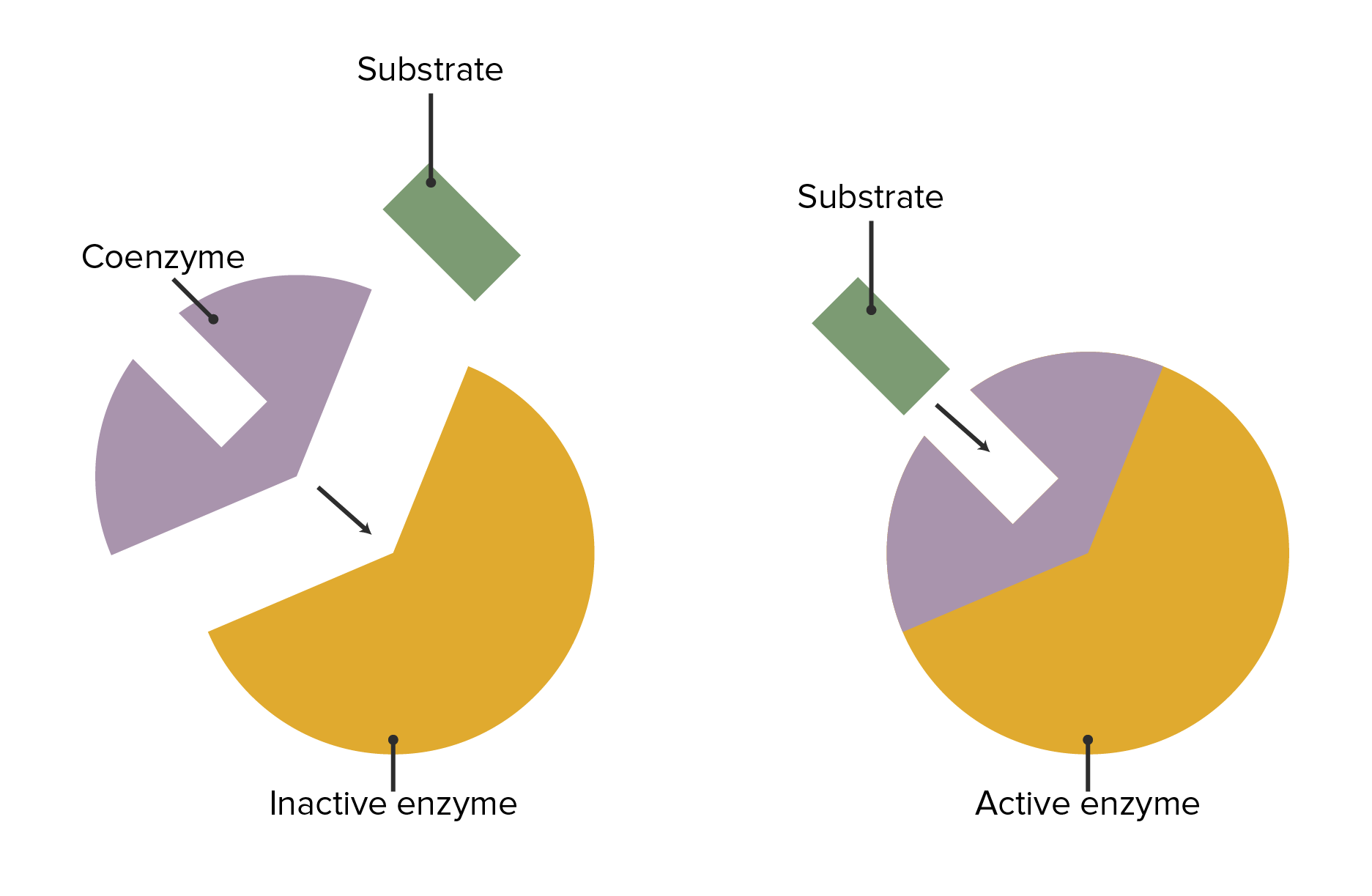
The interaction between enzymes, coenzymes, and substrate attachment
Image by Lecturio.| Coenzyme | Associated vitamin | Reaction type | Enzyme examples |
|---|---|---|---|
| Thiamine Thiamine Also known as thiamine or thiamin, it is a vitamin C12H17N4OSCl of the vitamin B complex that is essential to normal metabolism and nerve function and is widespread in plants and animals Water-soluble Vitamins and their Deficiencies pyrophosphate | B1 | Oxidative decarboxylation Decarboxylation The removal of a carboxyl group, usually in the form of carbon dioxide, from a chemical compound. Catabolism of Amino Acids |
|
| FAD/FADH2 | B2 | Electron transfer | Succinate dehydrogenase Succinate dehydrogenase A flavoprotein containing oxidoreductase that catalyzes the dehydrogenation of succinate to fumarate. In most eukaryotic organisms this enzyme is a component of mitochondrial electron transport complex II. Citric Acid Cycle |
| NAD NAD+ A coenzyme composed of ribosylnicotinamide 5′-diphosphate coupled to adenosine 5′-phosphate by pyrophosphate linkage. It is found widely in nature and is involved in numerous enzymatic reactions in which it serves as an electron carrier by being alternately oxidized (NAD+) and reduced (NADH). Pentose Phosphate Pathway+/NADP+ | B3 | Electron transfer | Many dehydrogenases |
| Lipoamide | B4 | Oxidative decarboxylation Decarboxylation The removal of a carboxyl group, usually in the form of carbon dioxide, from a chemical compound. Catabolism of Amino Acids | Pyruvate Pyruvate Derivatives of pyruvic acid, including its salts and esters. Glycolysis dehydrogenase |
| Coenzyme A (CoA) | B5 | Acyl group transfer | α-ketoglutarate dehydrogenase |
| Pyridoxal phosphate Phosphate Inorganic salts of phosphoric acid. Electrolytes | B6 | Transamination Transamination Transamination is the transfer of an amino group from an alpha-AA to an alpha-keto acid, which is an AA with an alpha-keto group (=O) instead of an alpha-amino group (NH2). Catabolism of Amino Acids | Alanine Alanine A non-essential amino acid that occurs in high levels in its free state in plasma. It is produced from pyruvate by transamination. It is involved in sugar and acid metabolism, increases immunity, and provides energy for muscle tissue, brain, and the central nervous system. Synthesis of Nonessential Amino Acids transaminase Transaminase A subclass of enzymes of the transferase class that catalyze the transfer of an amino group from a donor (generally an amino acid) to an acceptor (generally a 2-keto acid). Most of these enzymes are pyridoxyl phosphate proteins. Catabolism of Amino Acids ( ALT ALT An enzyme that catalyzes the conversion of l-alanine and 2-oxoglutarate to pyruvate and l-glutamate. Liver Function Tests) |
| Biotin | B7 | Carboxyl group transfer | Pyruvate carboxylase Pyruvate carboxylase A biotin-dependent enzyme belonging to the ligase family that catalyzes the addition of carbon dioxide to pyruvate. It is occurs in both plants and animals. Gluconeogenesis |
| Tetrahydrofolate Tetrahydrofolate Sulfonamides and Trimethoprim (THF) | B9 | Transfer of C1 groups |
|
| 5-deoxyadenosyl cobalamin Cobalamin A cobalt-containing coordination compound produced by intestinal microorganisms and found also in soil and water. Higher plants do not concentrate vitamin B 12 from the soil and so are a poor source of the substance as compared with animal tissues. Intrinsic factor is important for the assimilation of vitamin B 12. Folate and Vitamin B12 | B12 | Intramolecular rearrangements |
|
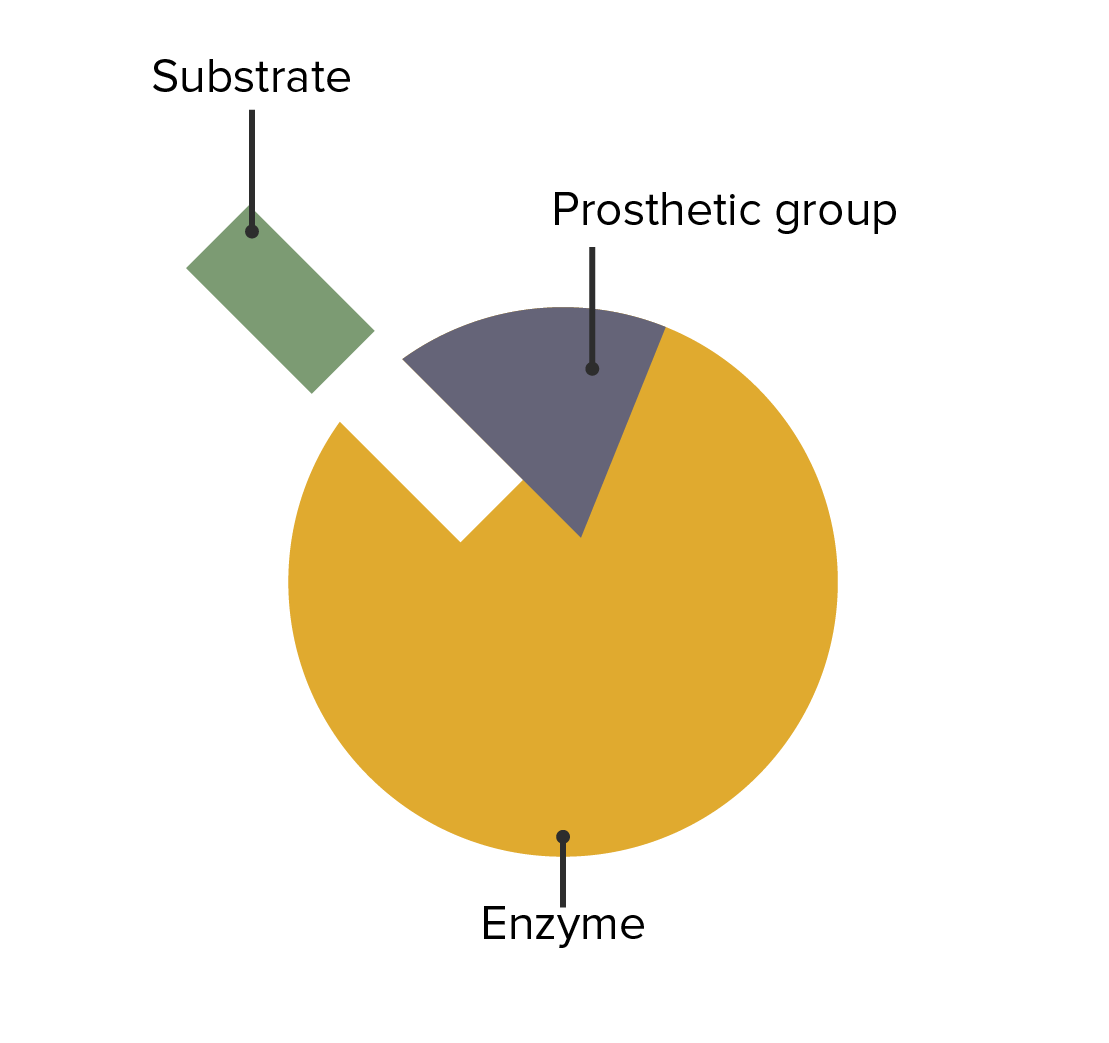
The interaction between enzymes, prosthetic groups, and substrate attachment
Image by Lecturio.The following conditions are caused by an enzymatic deficiency: